To the Editor:
Alagille Syndrome (AS) is a multisystemic genetic disorder, inherited through an dominant autosomal feature of variable expression. In 70% of cases, the causal gene is JAG 1, located in chromosome 20.1
Clinically, it appears with intermittent episodes of cholestasis that start in the neonatal period and are due to the absence of intrahepatic biliary ducts. Malformations associated with the syndrome include a peculiar phenotype, defects of the vertebral arches (butterfly-like vertebral defects), ocular anomalies (posterior embryotoxon) and cardiovascular anomalies. Of the latter, the most characteristic is the pulmonary artery condition that causes different degrees of subpulmonary stenosis.2
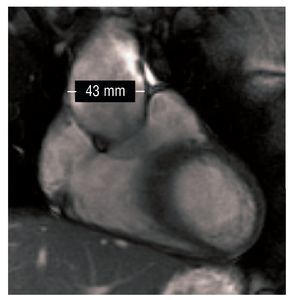
We present the case of a man aged 17 years diagnosed with AS who was referred for reexamination of a cardiopathy rejected in his infancy. The patient was completely asymptomatic. Physical examination revealed a mild protosystolic murmur over the left sternal border. The electrocardiogram showed no abnormalities of interest. Transthoracic echocardiography revealed a discrete dilatation of the ascending aorta from the root, of 42 mm in diameter (23 mm/m2) at the level of the Valsalva sinus. We identified mild aortic regurgitation over the apparently-functional tricuspid valve. Ventricular function was conserved and the rest of the study (valve and pulmonary artery) showed no abnormalities. Given these findings, we completed the examination with cardiac and major vessel resonance that revealed aortic dilatation limited to the lower thirds and half of the ascending aorta, where the aorta was 43 mm in diameter (24 mm/m2). At the level of the aortic arch and descending aorta, aortic caliber was normal (Figure 1).

Figure 1. Cardiovascular resonance sagittal slice: the aortic aneurysm is visible in the inferior third and half of the ascending aorta.
We hypothesize that an abnormal vascular formation may be involved in the principal mechanism of AS.3 The most extensive published review retrospectively studied 269 patients with AS and put prevalence of noncardiac vascular disease at 9%. Its presence has been associated with adverse prognosis. In the aforementioned series, 34% of patients who died presented some cardiovascular disease.4 The type and location of malformations reported elsewhere vary greatly: cerebral vascular anomalies,5,6 carotid aneurysm, hepatic venous system alterations8 and vasculorrenal disease9 have been described. The aortic condition has been described as descending aorta coarctation.10 Our patient is the first example of AS with a thoracic aorta aneurysm in a live patient that has been published. Previously this has only been described as an autopsy finding in three patients who died of sudden death.4
Due to our ignorance of its natural history, the most adequate therapeutic strategy has not been established. Bearing in mind that the only cases published were not diagnosed and that these patients were victims of sudden death, in order to indicate for surgery a reasonable attitude would be to apply criteria similar to those for connective tissue illnesses, entities with which AS apparently shares certain similarities.
In our patient, due to the size of the aneurysm, we decided on a wait-and-see policy with strict clinical and imaging control in order to consider a surgical intervention when there was evidence of significant growth.