El síndrome de Alagille (SA) es un trastorno genético multisistémico, heredado por rasgo autosómico dominante de expresión variable. En el 70% de los casos el gen causal es el JAG 1, localizado en el cromosoma 201.
Clínicamente se manifiesta con episodios intermitentes de colestasis que se inician en el período neonatal y se deben a la ausencia de conductos biliares intrahepáticos. Las malformaciones asociadas al síndrome incluyen un fenotipo peculiar, defectos de los arcos vertebrales (vértebras en mariposa), anomalías oculares (embriotoxon posterior) y anomalías cardiovasculares. De estas últimas, la más característica es la afección de la arteria pulmonar que origina diferentes grados de estenosis subpulmonar2.
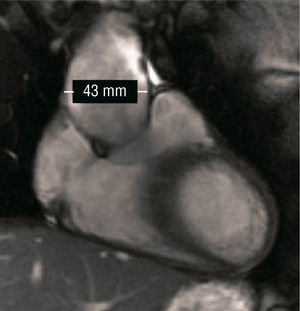
Presentamos el caso de un varón de 17 años diagnosticado de SA que fue remitido para reevaluar una cardiopatía que ya había sido descartada en la infancia. El paciente se encontraba completamente asintomático. Destacaba en la exploración física un ligero soplo protosistólico en borde esternal izquierdo. El electrocardiograma no mostraba anomalías de interés. El ecocardiograma transtorácico reveló una discreta dilatación de la aorta ascendente desde la raíz, que llegaba a 42 mm de diámetro (23 mm/m2) a nivel de los senos de Valsalva. Se apreciaba una regurgitación aórtica ligera sobre válvula trivalva de aspecto funcional. La función ventricular estaba conservada y el resto del estudio (válvula y arteria pulmonar) no mostraba alteraciones. Ante los hallazgos, se completó la valoración con una resonancia cardiaca y de grandes vasos, en la que se encontró que la dilatación aórtica quedaba limitada a los tercios inferior y medio de la aorta ascendente, donde la aorta alcanzaba 43 mm de diámetro (24 mm/m2). A nivel de cayado y aorta descendente, el calibre aórtico era normal (fig. 1).

Fig. 1. Corte sagital de resonancia cardiovascular: se visualiza el aneurisma aórtico en el tercio inferior y medio de la aorta ascendente.
Se ha postulado que una formación vascular anormal estaría involucrada en el mecanismo principal del SA3. La revisión publicada más extensa, que estudió de forma retrospectiva a 269 pacientes con SA, cifra en un 9% la prevalencia de afección vascular no cardiaca. Su presencia se ha relacionado con un pronóstico adverso. En la citada serie, el 34% de los fallecidos presentaba alguna afección cardiovascular4. El tipo y la localización de las malformaciones comunicadas en la bibliografía son muy diversos. Se han descrito anomalías vasculares cerebrales5,6, aneurisma carotídeo7, alteraciones del sistema venoso hepático8 y afección vasculorrenal9. La afección aórtica ha sido descrita en forma de coartación de aorta descendente10. El nuestro es el primer SA con un aneurisma de aorta torácica en un paciente vivo que se haya publicado. Tan sólo se describe como hallazgo necrópsico en tres casos que fallecieron de forma súbita4.
Debido al desconocimiento de su historia natural, no se ha establecido cuál es la estrategia terapéutica más adecuada. Teniendo en cuenta que los únicos casos publicados no estaban diagnosticados y causaron muerte súbita, para indicar la cirugía una actitud razonable sería aplicar criterios semejantes a los de las enfermedades del tejido conectivo, entidades con las que el SA parece compartir cierta similitud.
En nuestro caso, debido al tamaño del aneurisma, se decidió una actitud expectante con un estricto control clínico y de imagen para plantear una intervención quirúrgica cuando se evidencie un crecimiento significativo.