Glycogen storage diseases (GSD) may mimic hypertrophic cardiomyopathy (HCM).1 Some more recently identified GSD, also with heart involvement, are barely known and thus difficult to suspect (table 1). Herein we highlight that GSD type XV (OMIM #613507) can also present as myocarditis or even evoke a left ventricular (LV) arrhythmogenic cardiomyopathy.
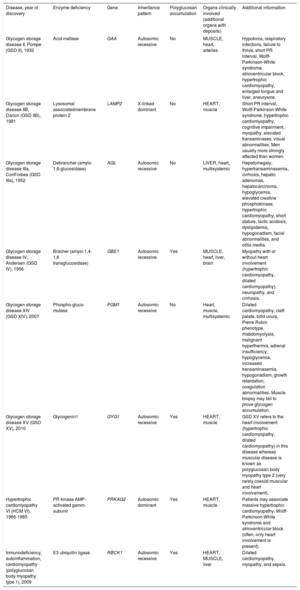
Summary of glycogen storage diseases with heart involvement
| Disease, year of discovery | Enzyme deficiency | Gene | Inheritance pattern | Polyglucosan accumulation | Organs clinically involved (additional organs with deposits) | Additional information |
|---|---|---|---|---|---|---|
| Glycogen storage disease II, Pompe (GSD II), 1932 | Acid maltase | GAA | Autosomic recessive | No | MUSCLE, heart, arteries | Hypotonia, respiratory infections, failure to thrive, short PR interval, Wolff-Parkinson-White syndrome, atrioventricular block, hypertrophic cardiomyopathy, enlarged tongue and liver, aneurysms. |
| Glycogen storage disease IIB, Danon (GSD IIB), 1981 | Lysosomal associatedmembrane protein 2 | LAMP2 | X-linked dominant | No | HEART, muscle | Short PR interval, Wolff-Parkinson-White syndrome, hypertrophic cardiomyopathy, cognitive impairment, myopathy, elevated transaminases, visual abnormalities. Men usually more strongly affected than women. |
| Glycogen storage disease IIIa, CoriForbes (GSD IIIa), 1952 | Debrancher (amylo-1,6-glucosidase) | AGL | Autosomic recessive | No | LIVER, heart, multisystemic | Hepatomegaly, hypertransaminasemia, cirrhosis, hepatic adenomas, hepatocarcinoma, hypoglycemia, elevated creatine phosphokinase, hypertrophic cardiomyopathy, short stature, lactic acidosis, dyslipidemia, hypogonadism, facial abnormalities, and otitis media. |
| Glycogen storage disease IV, Andersen (GSD IV), 1956 | Bracher (amylo 1,4-1,6 transglucosidase) | GBE1 | Autosomic recessive | Yes | MUSCLE, heart, liver, brain | Myopathy with or without heart involvement (hypertrophic cardiomyopathy, dilated cardiomyopathy), neuropathy, and cirrhosis. |
| Glycogen storage disease XIV (GSD XIV), 2007 | Phospho-gluco-mutase | PGM1 | Autosomic recessive | No | Heart, muscle, multisystemic | Dilated cardiomyopathy, cleft palate, bifid uvula, Pierre Robin phenotype, rhabdomyolysis, malignant hyperthermia, adrenal insufficiency, hypoglycemia, increased transaminasemia, hypogonadism, growth retardation, coagulation abnormalities. Muscle biopsy may fail to prove glycogen accumulation. |
| Glycogen storage disease XV (GSD XV), 2010 | Glycogenin1 | GYG1 | Autosomic recessive | Yes | HEART, muscle | GSD XV refers to the heart involvement (hypertrophic cardiomyopathy, dilated cardiomyopathy) in this disease whereas muscular disease is known as polyglucosan body myopathy type 2 (very rarely coexist muscular and heart involvement). |
| Hypertrophic cardiomyopathy VI (HCM VI), 1966-1985 | PR kinase AMP-activated gamm-subunit | PRKAG2 | Autosomic dominant | Yes | HEART, muscle | Patients may associate massive hypertrophic cardiomyopathy, Wolff-Parkinson-White syndrome and atrioventricular block (often, only heart involvement is present). |
| Inmunodeficiency, autoinflammation, cardiomyopathy (polyglucosan body myopathy type 1), 2009 | E3 ubiquitin ligase | RBCK1 | Autosomic recessive | Yes | HEART, MUSCLE, liver | Dilated cardiomyopathy, myopathy, and sepsis. |
Predominant organ involvement is depicted in capital letters.
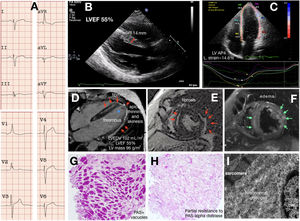
A young proband reported limiting chest pain, vague presyncopal spells, and progressive weakness. A mild increase in blood troponin T and urine protein levels were detected with normal creatinine phosphokinase. His electrocardiogram demonstrated sinus rhythm, atypical complete right bundle branch block, and left posterior hemiblock (figure 1A); isolated infrequent supraventricular and ventricular premature beats were noted by Holter and implanted loop recorder. Coronary stenosis was ruled out by coronary computed tomography angiography. Cardiac imaging by echocardiography and cardiac magnetic resonance imaging identified structural abnormalities limited to the left ventricle. Abnormal features included LV mild-to-moderate hypertrophy only at the basal septal segment, lower limit LV ejection fraction (LVEF), a slightly decreased global longitudinal strain, slightly increased volumes, thinned and hypokinetic apical and lateral walls with a laminar thrombus, severe epi/intramyocardial edema and scarring (figure 1B-F). Myocarditis was diagnosed and the patient was put on colchicine, bisoprolol, and oral anticoagulants. The patient soon had a cerebellar stroke with an ad integrum recovery but otherwise symptoms and tests have remained unchanged during the 24-month follow-up. Persisting chest pain prompted an endomyocardial biopsy, which showed no disarray or abnormal fibrosis and revealed a severe myocardial vacuolization peripherally displacing the nuclei. The marked periodic acid-Schiff (PAS) positivity at the vacuoles gave the diagnosis of GSD and its partial attenuation if pretreatment with diastase was included in the staining protocol and was consistent with polyglucosan deposits (figure 1G-H). Ultrastructural images also fitted with the diagnosis (figure 1I). Next generation sequencing (NextSeq 500, Illumina Technologies) was used to assess for GSD, HCM, and arrhythmogenic cardiomyopathy genes. The proband carried 2 already published pathogenic variants in the GYG-1 gene, namely the p.Asp102His and the p.Gly135Arg.2 Skeletal muscular involvement was further ruled out with a thorough clinical examination by an expert neurologist, a whole-body computed tomography, standard respiratory function tests, and an exercise test. The need for anticoagulation prevented us from performing a muscle biopsy. Regarding treatment, colchicine and beta-blockers were withdrawn and anticoagulation intensified. The 6 relatives of the 3-generation family study were phenotype negative and heterozygous for only 1 of the mutations.
. The asterisk highlights the apical thrombus. The green arrows point to the myocardial edema. G-H: histology. I: ultrastructural characterization. IVS, interventricular septum thickness; LVEF, left ventricular ejection fraction; LV AP4 L strain, left ventricular apical 4-chamber longitudinal strain; LVEDV, left ventricular end-diastolic volume; LV, left ventricular; PAS, periodic acid-Schiff.")
Cardiac GSD type XV phenotype. A: electrocardiogram. B-C: echocardiography. D-F: cardiac magnetic imaging. The red arrows point to the late gadolinium enhancement (fibrosis). The asterisk highlights the apical thrombus. The green arrows point to the myocardial edema. G-H: histology. I: ultrastructural characterization. IVS, interventricular septum thickness; LVEF, left ventricular ejection fraction; LV AP4 L strain, left ventricular apical 4-chamber longitudinal strain; LVEDV, left ventricular end-diastolic volume; LV, left ventricular; PAS, periodic acid-Schiff.
The term GSD type XV is preferred for the cardiomyopathic phenotype while polyglucosan body myopathy type 2 (PGBM2, OMIM #616199) applies for an allellic disease characterized by late onset and slowly progressive myopathy without cardiac involvement.2–5 Thirty-eight patients with PGBM2 have been described so far and ours is the fifth case of GSD type XV.2–4 All these patients harbor 2 GYG-1 mutations and polyglucosan accumulation is identified either in cardiomyocytes or the skeletal muscle. Of note, nonsense mutations are more common among myopathic patients than in cardiomyopathic patients.3,5
Having reviewed the available evidence3,4 and our proband, we stress that GSD type XV represents a cardiological diagnosis, with microscopic biventricular involvement, although cardiac imaging only detects an isolated LV disease. Of note, this disease may not necessarily exhibit massive LV hypertrophy (14-23 mm in the 5 patients so far identified) and has never been associated with cognitive impairment, atrioventricular block or pre-excitation, as have other well-known GSD, such as PRKAG2, Danon and Pompe diseases.1 Instead, long-lasting chest pain (80%), LV regional abnormalities (60% thinning, 40% hypo/akinesis), LV tissue characterization alterations (100% scarring, 20% edema), and systemic embolic events (40% stroke, 20% LV thrombus) are the most representative features. Additionally, there have been reports of a wide range of LV dysfunction (LVEF 25%-55%, slightly-severely increased volumes, 20% need for heart transplant) associated with life-threatening ventricular tachyarrhythmias (40%), and rarely skeletal myopathy (20%). Of interest, the disease is fully penetrant in men but not in women (a woman homozygous for the p.Asp102His mutation did not show any signs or symptoms on heart and skeletal muscle assessment2).
Finally, we stress that GSD type XV should be considered as a phenocopy not only of HCM but also of myocarditis and LV dominant arrhythmogenic cardiomyopathy (especially if the left ventricular hypertrophy is only mild and the LV scarring is pronounced). Notably, the endomyocardial biopsy and histological studies are crucial to direct the genetic assessment. If suspected, cardiologists should ensure that GYG-1 is included in the panel selected for the genetic study. Unfortunately, there is currently no treatment available for disease management. Arrhythmias and heart failure are managed symptomatically.
FundingThis work was partially supported by grants from the Instituto de Salud Carlos III, FEDER Unión Europea, Una forma de hacer Europa [PI18/01582] and La Fe Biobank [PT17/0015/0043] and Memorial Nacho Barberá.