




Myocarditis is defined as myocardial inflammation and its etiology is highly diverse, including infectious agents, drugs, and autoimmune diseases. The clinical presentation also varies widely, extending beyond the classic clinical picture of acute chest pain, and includes cases of cardiomyopathy of unknown cause whose etiology may be inflammatory. Because certain patients may benefit from targeted treatments, the search for the etiology should begin when myocarditis is first suspected. There remain several areas of uncertainty in the diagnosis and treatment of this disease. Consequently, this consensus document aims to provide clear recommendations for its diagnosis and treatment. Hence, a diagnostic algorithm is proposed, specifying when non-invasive diagnosis with cardiac MR is appropriate vs a noninvasive approach with endomyocardial biopsy. In addition, more novel aspects are discussed, such as when to suspect an underlying genetic etiology. The recommendations cover the management of myocarditis and inflammatory cardiomyopathy, both for general complications and specific clinical entities.
Keywords
Myocarditis is an etiologically diverse inflammatory disease involving the heart muscle. Its multiple causes include infections, toxins, and autoimmune diseases.1–3 Diagnosis can be challenging due to the variability in clinical presentations and the lack of accurate diagnostic tools. Treatment options are also limited, contingent on the cause, and subject to uncertainty. The Myocarditis Working Group of the Spanish Society of Cardiology (SEC) has prepared a consensus statement on the diagnosis and treatment of myocarditis and inflammatory cardiomyopathy to address the need for clear recommendations and standardized clinical practices in this setting. A small group of authors and 2 coordinators were chosen from among the members of the working group and tasked with preparing a draft statement and reaching agreement on the recommendations to be included over the course of several meetings. The resulting statement was revised and subsequently approved by all the members of the working group.
A summary of the various definitions of myocarditis is provided in table 1.1–3
Definitions
| Clinically suspected myocarditisSuggestive clinical symptoms and at least 1 objective abnormality in:a) Basic tests (suggestive electrocardiographic or echocardiographic changes)b) Myocardial injury biomarkersc) Suggestive signs on CMR, but not sufficient for a firm diagnosis to be established |
| Confirmed myocarditisClinically suspected myocarditis plus at least 1 of the following:a) CMR criteria (noninvasive diagnosis):• At least 1 T1-based criterion (abnormal native T1, increased extracellular volume, or late gadolinium enhancement) and at least 1 T2-based criterion (increased T2 signal intensity or abnormal T2 mapping)b) EMB-proven histologic or immunohistologic criteria (invasive diagnosis)• Histologic criteria (Dallas): histologic evidence of inflammatory infiltrates in the myocardium, associated with nonischemic necrosis• Immunohistologic criteria: ≥ 14 leukocytes/mm2 including up to 4 monocytes/mm2, with the presence of > 7 CD3+ cells (T lymphocytes) per mm2• Borderline myocarditis: histologic evidence of inflammatory infiltrates, but without necrosis |
| Fulminant myocarditisInflammatory myocardial disease characterized by acute heart failure and hemodynamic instability (<2-4 weeks since symptom onset). The EMB shows lymphocytic foci. Fulminant myocarditis should not be confused with other forms potentially associated with hemodynamic instability that have specific treatments, such as giant cell myocarditis, eosinophilic myocarditis, and sarcoidosis |
| Acute vs chronic myocarditisAcute myocarditisTime from onset of symptoms to diagnosis <1 mo - Complicated: LVEF <50%, sustained ventricular arrhythmias, severe conduction disorders, heart failure, or cardiogenic shock- Uncomplicated: absence of criteria for complicated acute myocarditisChronic myocarditisTime from onset of symptoms to diagnosis >1 mo |
| Inflammatory cardiomyopathyChronic cardiomyopathy associated with ventricular dysfunction (LVEF <50%) |
CMR, cardiac magnetic resonance imaging; EMB, endomyocardial biopsy; LVEF, left ventricular ejection fraction.
Recommendation. A high index of diagnostic suspicion should be maintained for myocarditis due to its variable clinical presentation.
Common manifestations in the weeks preceding acute myocarditis include fever and other prodromal symptoms indicative of a viral infection, such as skin rash and oropharyngeal, respiratory, and gastrointestinal symptoms.4 The most likely clinical presentations are listed below:
- a)
Asymptomatic presentation.Asymptomatic myocarditis is detected following observation of other abnormalities, in particular, electrocardiogram (ECG) changes such as repolarization patterns and premature ventricular contractions.5
- b)
Chest pain similar to that observed in pericarditis and acute coronary syndrome. Chest pain is the most common manifestation of myocarditis and tends to be accompanied by ST-T-wave changes and persistently elevated troponins.2,3
- c)
Dyspnea or acute or subacute heart failure (onset < 1-3 months2,3). These are the second most common presentations of myocarditis and are accompanied by altered contractility, with variable increases in thickness and normal or slightly dilated ventricles.6
- d)
Palpations, syncope, and arrhythmias. Arrhythmias in myocarditis range from inappropriate sinus tachycardia to frequent (typically ventricular) premature contractions, sustained tachyarrhythmias, new-onset conduction disorders, and bradyarrhythmias. They can also cause sudden cardiac death or be diagnosed postmortem.
- e)
Established cardiomyopathy (> 3 months2,3). Onset and progression of myocarditis can sometimes go unnoticed. At diagnosis, myocarditis will have progressed to cardiac dysfunction and cardiomyopathy, with normal or abnormal troponin levels and variable degrees of biventricular dilation and dysfunction.
Recommendation. Diagnostic testing should start as soon as myocarditis is suspected.
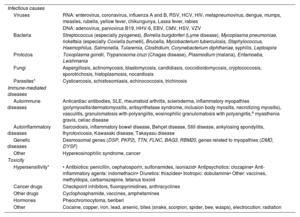
Underlying causes should be investigated as soon as myocarditis is suspected. Potential causes should be contemplated when taking the patient's history and ordering tests, as their identification will determine treatment. These causes are listed in table 2.2,3,6-8 Although most cases of myocarditis are thought to be of viral origin,2,3,6-8 a definitive diagnosis is seldom reached. Where possible, it is important to identify the virus involved, as this will determine the diagnostic process and treatment.
Causes of myocarditis2,3,6-8
| Infectious causes | |
| Viruses | RNA: enterovirus, coronavirus, influenza A and B, RSV, HCV, HIV, metapneumovirus, dengue, mumps, measles, rubella, yellow fever, chikungunya, Lassa fever, rabies |
| DNA: adenovirus, parvovirus B19, HHV-6, EBV, CMV, HSV, VZV | |
| Bacteria | Streptococcus (especially pyogenes), Borrelia burgdorferi (Lyme disease), Mycoplasma pneumoniae, rickettsia (especially Coxiella burnetii), Brucella, Mycobacterium tuberculosis, Staphylococcus, Haemophilus, Salmonella, Tularemia, Clostridium, Corynebacterium diphtheriae, syphilis, Leptospira |
| Protozoa | Toxoplasma gondii, Trypanosoma cruzi (Chagas disease), Plasmodium (malaria), Entamoeba, Leishmania |
| Fungi | Aspergillosis, actinomycosis, blastomycosis, candidiasis, coccidioidomycosis, cryptococcosis, sporotrichosis, histoplasmosis, nocardiosis |
| Parasites* | Cysticercosis, schistosomiasis, echinococcosis, trichinosis |
| Immune-mediated diseases | |
| Autoimmune diseases | Anticardiac antibodies, SLE, rheumatoid arthritis, scleroderma, inflammatory myopathies (polymyositis/dermatomyositis, antisynthetase syndrome, inclusion body myositis, necrotizing myositis), vasculitis, granulomatosis with polyangiitis, eosinophilic granulomatosis with polyangiitis,a myasthenia gravis, celiac disease |
| Autoinflammatory diseases | Sarcoidosis, inflammatory bowel disease, Behçet disease, Still disease, ankylosing spondylitis, thyrotoxicosis, Kawasaki disease, Takayasu disease |
| Genetic diseases | Desmosomal genes (DSP, PKP2), TTN, FLNC, BAG3, RBM20, genes related to myopathies (DMD, DYSF) |
| Other | Hypereosinophilic syndrome, cancer |
| Toxicity | |
| Hypersensitivity* | • Antibiotics: penicillin, cephalosporin, sulfonamides, isoniazid• Antipsychotics: clozapine• Anti-inflammatory agents: indomethacin• Diuretics: thiazides• Inotropic: dobutamine• Other: vaccines, methyldopa, carbamazepine, tetanus toxoid |
| Cancer drugs | Checkpoint inhibitors, fluoropyrimidines, anthracyclines |
| Other drugs | Cyclophosphamide, vaccines, amphetamines |
| Hormones | Pheochromocytoma, beriberi |
| Other | Cocaine, copper, iron, lead, arsenic, bites (snake, scorpion, spider, bee, wasps), electrocution, radiation |
CMV, cytomegalovirus; DNA, deoxyribonucleic acid; EBV, Epstein-Barr virus; HCV, hepatitis C virus; HHV-6, human herpesvirus 6; HIV, human immunodeficiency virus; HSV, herpes simplex virus; RNA, ribonucleic acid; RSV, respiratory syncytial virus; SLE, systemic lupus erythematosus; VZV, varicella-zoster virus
Recommendation. All patients with suspected myocarditis should undergo ECG, laboratory tests, and echocardiography.
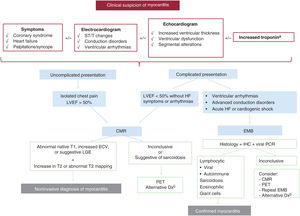
The recommended diagnostic algorithm for suspected myocarditis is shown in figure 1. Clinical suspicion is the first step.
.")
Diagnostic algorithm for suspected myocarditis. CMR, cardiac magnetic resonance; Dx, diagnosis; EBM, endomyocardial biopsy; ECV, extracellular volume; IHC, immunohistochemistry; LGE, late gadolinium enhancement; LVEF, left ventricular ejection fraction; PCR, polymerase chain reaction; PET, positron emission tomography.
a Normal troponin levels do not necessarily exclude a diagnosis of chronic myocarditis or suspected inflammatory cardiomyopathy.
b Consider a genetic study (together with the recommendations in figure 2).
ECG findings, while often nonspecific, are altered in most cases of myocarditis, and normal findings do not exclude a diagnosis.3 The most common finding is ST elevation in the inferolateral leads or in all leads (diffuse ST elevation), but other changes include repolarization abnormalities, pathologic Q waves, T-wave abnormalities, prolonged QRS duration, blocks, and frequent premature contractions.3 Advanced atrioventricular block should raise suspicion of cardiac sarcoidosis, Lyme disease, or immunotherapy-associated myocarditis.2 Low voltages (due to myocardial edema), ventricular arrhythmias, and conduction disorders should raise suspicion of high-risk myocarditis with a potentially fulminant course.8
Laboratory testsRecommendation. The following laboratory tests should be ordered on suspicion of myocarditis.
- •
High-sensitivity cardiac troponin. Troponin levels are usually elevated in acute myocarditis and can be consistently raised or rise and fall. They are only weakly correlated with disease severity. Negative findings do not rule out a diagnosis.3
- •
N-terminal pro-B-type natriuretic peptide or brain natriuretic peptide (related to heart failure severity).
- •
Inflammatory markers (C-reactive protein and erythrocyte sedimentation rate). These markers are mostly positive. Persistently high levels should increase suspicion of an underlying autoimmune disease.2
- •
Complete blood count. Observation of eosinophilia points to a possible case of eosinophilic myocarditis.9
- •
Biochemistry. Liver function, kidney function, electrolytes, thyroid profile, and creatine kinase.
A recently described circulating microRNA (hsa:Chr8:96) was found to distinguish between myocarditis and acute coronary syndrome.10 This marker has the potential to facilitate early diagnosis of myocarditis, but has yet to be applied in clinical practice.
Targeted investigations should be performed when specific causes are suspected (table 1 of the supplementary data). If there are no clear causes but autoimmune myocarditis is strongly suspected, antiheart autoantibodies, anti-intercalated disk autoantibodies, and anti-β-1 adrenergic receptor antibodies should be assessed when available.3
EchocardiogramRecommendation. An echocardiogram should be performed as soon as myocarditis is clinically suspected. Testing is urgent in hemodynamically unstable patients. A repeat echocardiogram should be contemplated after a few days, particularly in patients with an unfavorable clinical course.
Early assessment of ventricular function is helpful for determining level of care and prognosis. In the early course of myocarditis, approximately 75% of patients will have a normal-sized left ventricle and normal systolic function.1 Because heart function can deteriorate rapidly (within days of onset), a repeat echocardiogram should be performed if any clinical changes are observed. Echocardiographic findings suggestive of myocarditis include increased myocardial thickness and granular echogenicity, both linked to the presence of edema. Other common findings are segmental contractility changes, in particular hypokinesia in the inferior and inferolateral regions of the left ventricle, and varying degrees of pericardial effusion.1 Myocardial deformation measured by speckle-tracking echocardiography is predictive of late gadolinium enhancement (LGE) in cardiac magnetic resonance imaging (CMR)11; it is reduced in affected segments.
Cardiac magnetic resonance imagingRecommendation. Compatible symptoms together with objective evidence of abnormalities in standard tests (echocardiogram, laboratory tests, and ECG) support a suspected diagnosis of myocarditis and justify CMR.
Recommendation. When acute myocarditis is suspected, CMR should be performed rapidly (ideally within 7 days) and include T1/T2mapping to help establish a confirmatory diagnosis. CMR is an alternative to endomyocardial biopsy (EMB), but it cannot establish a cause.
CMR is recommended in all patients with clinical suspicion of myocarditis and who have elevated biomarkers or ECG or echocardiographic changes indicative of myocardial injury (see section “Assessment of cardiac anatomy”).
The updated Lake Louise criteria from 2018 are based on a combination of CMR images used to assess the presence of myocardial edema and fibrosis. A diagnosis of myocarditis is based on the detection of at least 1 T1-based criterion (abnormal native T1, increased extracellular volume, or LGE) and at least 1 T2-based criterion (increased T2 signal intensity or abnormal T2 mapping) on CMR. Concomitant pericarditis and global or regional systolic dysfunction also support a diagnosis of myocarditis. CMR should be performed within 2 weeks of the onset of clinical symptoms, and ideally within 7 days of suspicion.12 Advanced imaging techniques such as feature tracking (to assess myocardial deformation) and texture analysis (radiomics) have shown promising early results.13 The addition of parametric mapping techniques has been found to improve the diagnostic yield of CMR in myocardial inflammation (from 84% to 90%-96%).14
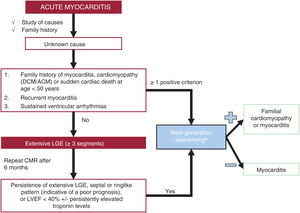
Genetic testingThe indications for genetic testing are summarized in figure 2.
: ACTC1, BAG3, DES, DSC2, DSG2, DSP, EMD, FLNC, JUP, MYBPC3, MYH7, PKP2, PLN, TMEM43, LMNA, RBM20, SCN5A, TNNC1, TNNI3, TNNT2, TPM1, TTN.")
Recommendations for genetic testing in myocarditis. ACM, arrhythmogenic cardiomyopathy; CMR, cardiac magnetic resonance; DCM, dilated cardiomyopathy; LGE, late gadolinium enhancement; LVEF, left ventricular ejection fraction.
aGenes that should be prioritized (in alphabetical order): ACTC1, BAG3, DES, DSC2, DSG2, DSP, EMD, FLNC, JUP, MYBPC3, MYH7, PKP2, PLN, TMEM43, LMNA, RBM20, SCN5A, TNNC1, TNNI3, TNNT2, TPM1, TTN.
Recommendation. EMB with immunohistochemical staining should be performed on suspicion of a specific cause that might benefit from targeted treatment.
EMB is the only test that can establish a definitive cause of myocarditis and, accordingly, enable initiation of targeted treatment. Immunohistochemical analysis is recommended, as it significantly increases the sensitivity of EMB compared with histologic examination only (Dallas criteria).15,16 The indications for immunohistochemistry are shown in table 3.
Clinical indications for endomyocardial biopsy
| Acute forms |
| Suspected fulminant myocarditis or acute myocarditis with left ventricular dysfunction + acute heart failure and/or rhythm disorders (high-grade atrioventricular block, ventricular arrhythmias refractory to treatment) |
| Suspected immune checkpoint inhibitor-mediated cardiotoxicity |
| Suspected eosinophilic myocarditis |
| Chronic forms |
| Progressive heart failure unresponsive to treatment ± rhythm disorders in patients with underlying autoimmune diseases and potential cardiac involvement |
| Ventricular dysfunction with refractory heart failure after at least 3 mo of standard treatment and after ruling out other causes of systolic dysfunction |
The current indications for EMB according to various international scientific societies are listed in table 2 of the supplementary data.2,17,18
The technical specifications for EMB are summarized in table 3 of the supplementary data.
Fluoro-[18F]-deoxy-2-D-glucose positron emission tomographyRecommendation. Fluoro-[18F]-deoxy-2-D-glucose positron emission tomography (FDG PET) should only be used for diagnosis or to assess treatment response when CMR and EMB are unavailable or inconclusive.
PET provides metabolic information about myocardial inflammation, detected as an increase in FDG uptake. Adequate preparation in the 36 to 72 hours preceding the scan is needed to suppress normal glucose metabolism (table 3 of the supplementary data). PET is an alternative diagnostic tool for stable patients who are unable to undergo CMR (because they have an implantable device, for example) or who have inconclusive results on other tests. It is also very useful for diagnosing and monitoring sarcoidosis19 and investigating autoimmune-mediated inflammation in other organs.
Compared with EMB, FDG PET has high sensitivity (75%) and specificity (67%) for suspected myocarditis.20 It can also be used to monitor changes to myocardial injury, assess responses to immunosuppressive agents, and guide the withdrawal of these agents during follow-up.20 The combined use of PET and CMR is currently being investigated, as it may be more effective than either technique in isolation.21
Assessment of cardiac anatomyRecommendation. The possibility of coronary artery disease must be considered and ruled out using coronary computed tomography (CT) angiography (low or intermediate probability) or coronary angiography (high probability).
Early invasive angiography is the diagnostic test of choice for adults with known cardiovascular risk factors, chest pain, ECG changes indicative of ischemia, clinical instability, acute troponin elevation, or regional contractile abnormalities on the echocardiogram. Coronary CT, however, should be prioritized in young, hemodynamically stable, patients without risk factors when there is a low to intermediate suspicion of ischemia. CT can also be used to rule out conditions such as coronary anomaly and dissection. A cardiac anatomy assessment is not needed when there is a low index of suspicion for ischemia and when early CMR shows findings consistent with myocarditis.
TREATMENT OF MYOCARDITISGeneral measuresRecommendation. All patients with probable or clinically suspected acute myocarditis should be admitted to hospital.
Rest and clinical observation are essential during the early stages of myocarditis, as acute forms have an uncertain course. Hospital admission is therefore recommended to confirm the diagnosis, investigate underlying causes, and determine whether the presentation is complicated or uncomplicated.
Bedside monitoring of vital signs and ECG are recommended for patients on general wards without evidence of severe disease. Patients with suspected myocarditis and ventricular arrhythmias, conduction disorders, impaired ventricular function, or heart failure should be admitted to an acute care unit.
General treatment varies according to whether the presentation is classified as complicated or uncomplicated. The recommended treatment for uncomplicated myocarditis (isolated chest pain and left ventricular ejection fraction [LVEF] >50%) is monitoring and conventional pain relief (paracetamol or metamizole). Use of nonsteroidal anti-inflammatory steroids (NSAIDs) in uncomplicated acute myocarditis has been debated and is not systemically recommended.3 The recommendation to avoid NSAIDs is based on animal studies from the 1980s and 1990s showing deleterious effects, such as increased viral loads and mortality.22 NSAIDs, however, could be considered in patients with persistent pain or concomitant pericardial involvement, as they have recently been shown to be safe in this setting.23
Colchicine is recommended in patients with associated pericardial involvement.24
Supportive treatment of complicated myocarditis is described in the section on the treatment of complications in the supplementary data. The treatments for specific forms of myocarditis are summarized in table 42,25-37 and in the supplementary data.
Immunosuppressive therapy for different forms of myocarditis
| Drug | Action | Treatment dose and duration | |||||
|---|---|---|---|---|---|---|---|
| Giant cell myocarditis | Fulminant lymphocytic myocarditis | Chronic lymphocytic myocarditis | Eosinophilic myocarditis | Sarcoidosis | ICI-mediated myocarditis | ||
| Corticosteroids | Suppresses leukocyte migration | Methylprednisolone IV or 500-1000 mg for 3 d; then, 1 mg/kg/24 h with gradual tapering25 | *Methylprednisolone IV, 7-14 mg/kg or 500-1000 mg for 3 d; then, 1 mg/kg/24 h with gradual tapering25 | *Methylprednisolone IV, 1 mg/kg/24 h for the first 4 wk followed by gradual tapering2 | *Methylprednisolone IV, 1 mg/kg/24 h for the first 4 wk followed by gradual tapering | *Prednisone 0.5 mg/kg for 1 mo; then gradual tapering over at least 12 mo26 | *Methylprednisolone IV 1000 mg for 3 d; then, 1 mg/kg/24 h with gradual tapering27 |
| Cyclosporine | Inhibits T-cell activation induced by interleukin 2 | Duration: indefinite25,28,29Target concentrations:0-3 mo: 150-250 ng/mL4-12 mo: 100-150 ng/mL>12 mo: 80-100 ng/mL | |||||
| Azathioprine | Inhibits purine synthesis, affecting DNA production in T and B cells | Duration: 1 y2 1-2 mg/kg/d divided into 2 doses | Duration: 6 mo1-2 mg/kg/d divided into 2 doses30 | Duration: 6 mo1-2 mg/kg/d divided into 2 doses | |||
| Methotrexate | Inhibits dihydrofolate reductase, the enzyme responsible for converting folic acid to tetrahydrofolate Inhibits proliferation and induces apoptosis of activated T lymphocytes | Second-line treatment if inflammation persists despite corticosteroid treatmentInitial oral/subcutaneous dose of 10-15 mg/wk, with a 5-mg increment every 2 wk to reach a dose of 20 mg31 | |||||
| Tacrolimus | Inhibits calcineurin-mediated T cell activation | Duration: indefinite2Target concentrations: 0-6 mo: 10-150 ng/mL>6 mo: 5-100 ng/mL | |||||
| Mycophenolate mofetil | Inhibits purine synthesis and selectively affects DNA production in T and B cells | Duration: 1 y, 500-1000 mg/12 h | Second-line treatment if inflammation persists despite corticosteroid treatment | ||||
| GTM | Polyclonal anti-T cell antibody | • Rabbit GTM: 100 mg/24 h or 1 mg/kg for 3-5 d32• Equine GTM: 500 mg on day 1, 250 mg on days 2-3, or 10 mg/kg every 3-4 d33 | Rabbit GTM27:1.5 mg/kg on day 1,0.5-1.5 mg/kg on days 2-6 (monitoring based on CD3 levels) | ||||
| Alemtuzumab | Monoclonal antibody that binds to CD52 on B and T cells, macrophages, monocytes, and natural killer cells | 30 mg in a single dose or two 15-mg doses2 | 30 mg in a single dose | ||||
| Immunoglobulins | Antigen-specific activity, multiple immunomodulatory activity | 2 g/kg in continuous infusion over 24-48 h or divided over 4 d. More experience in children34 | |||||
| Interferon beta | Antiviral and immunomodulatory activity mediated by cell receptors | 4×106 IU SC/48 h wk 18×106 IU from wk 2 to >6 mo | |||||
| Rituximab | Cytotoxicity due to antibodies against CD20+ B cells | 375 mg/m2/wk IV for 4 wk35 | 375 mg/m2/wk for 4 wk36 | ||||
| Infliximab/adalimumab | Tissue necrosis factor inhibitors | Third-line treatment if no response to corticosteroids or other immunosuppressive agents37 | |||||
| Imatinib | Inhibits tyrosine kinase activity of BCR-ABL, c-kit, and PDGFR proteins | Hypereosinophilic syndrome (myeloproliferative variant) |
GTM, antithymocyte globulin; IV, intravenous; NK, natural killer; PDGFR, platelet-derived growth factor receptor; SC, subcutaneous.
Recommendation. There is insufficient evidence to guide specific recommendations on the treatment of heart failure in myocarditis. Guideline recommendations for heart failure due to other causes should be followed.
Although no studies have specifically analyzed heart failure in myocarditis, the general consensus is to extrapolate clinical trial results and heart failure guideline recommendations38 regarding drugs with proven survival benefits, in particular quadruple therapy. Specific evidence is also lacking on how to treat patients with transient systolic dysfunction and recovered LVEF, but the recommendation is to maintain treatment, especially when CMR shows persistent LGE.39
Cardiogenic shockRecommendation. Rapid access to circulatory support should be available for patients with early signs of cardiogenic shock.
There is no evidence supporting the use of any specific treatments for cardiogenic shock in myocarditis. Patients with severe heart failure requiring inotropic agents should receive early care in an acute care unit with immediate access to mechanical circulatory support systems, such as ventricular assist devices (VADs) and extracorporeal membrane oxygenation (ECMO).40,41
Because cardiogenic shock is potentially reversible, the main purpose of treatment is to facilitate biventricular unloading, adequate systemic and coronary perfusion, and venous decongestion to prevent multiorgan dysfunction and provide a bridge to recovery or, if necessary, transplantation or long-term assist devices. These devices range from venoarterial ECMO to VADs with rotary or axial pumps. ECMO combined with devices that reduce left ventricular afterload and intracavitary pressures (eg, Impella; Abiomed, USA) appears to provide better myocardial recovery than ECMO alone.42-44
Right ventricular function and local expertise should be taken into account when choosing a device. Long-term VAD placement or heart transplantation should be considered in patients who cannot be weaned off mechanical circulatory support after 2 to 3 weeks.45
ArrhythmiasRecommendation. Arrhythmias may be reversible in the acute stages of myocarditis, and this possibility should be considered when taking decisions. Sarcoidosis and giant cell myocarditis are the most likely diagnoses in patients with myocarditis and ventricular arrhythmias.
Although ventricular arrhythmias are generally uncommon in myocarditis,3,46 they can occur in both acute and chronic forms, and particularly in cardiac sarcoidosis47 and giant cell myocarditis.48 Pharmacological treatment and close observation are recommended during acute stages. Invasive treatments should generally be avoided, as arrhythmias are typically caused by inflammation and often improve after its resolution. Beta-blockers can be useful for treating premature ventricular contractions or other arrhythmias.49 Supraventricular arrhythmias predominate in patients with less severe inflammation (ventricular arrhythmias are less common).50 Bradyarrhythmias are also less common, but can occur, particularly in Chagas disease, giant cell myocarditis, and autoimmune diseases with myocardial involvement.51
Residual fibrosis typically serves as the substrate for arrhythmias in chronic or late-stage myocarditis.52,53 General clinical practice guidelines should be followed, as specific recommendations are lacking.54
The value of implantable cardioverter-defibrillators (ICDs) has been debated, as most episodes of acute myocarditis are transient and potentially reversible. Secondary prevention ICDs can generally be considered following cardiac arrest due to ventricular fibrillation or in patients with symptomatic ventricular tachycardia, especially if arrhythmias persist beyond the acute stage of the disease despite pharmacological treatment.54 Consideration should always be given to the presence, extent, and potential reversibility of left ventricular dysfunction and the use of bridging strategies such as wearable defibrillator vests,55 particularly in primary prevention.
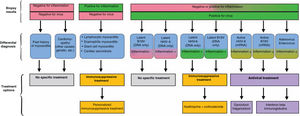
SPECIFIC FORMS OF MYOCARDITIS AND TREATMENTAcute lymphocytic myocarditisRecommendation. Corticosteroids should only be used to treat fulminant lymphocytic myocarditis or complicated acute myocarditis in patients awaiting EMB, the results of which should dictate treatment (figure 3).

Acute lymphocytic myocarditis is histologically characterized by a patchy inflammatory infiltrate with a predominant lymphocytic component and absent giant cells and granulomas. Eosinophils and neutrophils are common in nonchronic forms.56 Acute lymphocytic myocarditis is the least specific form of myocarditis and is mostly caused by viruses. Identification of a virus, however, does not necessarily imply causality, as viral infections are common and polymerase chain reaction tests are highly sensitive. Treatment is supportive and may include empirical corticosteroids for fulminant myocarditis or complicated cases pending EMB (table 4). The MYTHS clinical trial (NCT05150704) is currently evaluating the use of high-dose corticosteroids in complicated myocarditis with heart failure or cardiogenic shock.
Giant cell myocarditisRecommendation. Giant cell myocarditis should be treated aggressively and rapidly using combination immunosuppressive therapy: access to circulatory support must be ensured given the uncertain course of this disease.
Giant cell myocarditis is rare and has a very poor prognosis. Its rarity limits the feasibility of randomized trials. The little evidence that is available on giant cell myocarditis comes from observational studies (mostly retrospective) and cases series.25,29
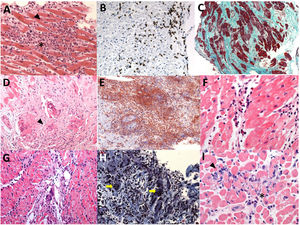
Histologically, giant cell myocarditis is characterized by lymphocytic infiltration, mononuclear cells, increased macrophages, and numerous giant cells. Giant cells are pathognomonic and must be present for a diagnosis to be made (figure 4). Giant cell myocarditis is the most severe form of myocarditis and tends to be associated with a higher diagnostic yield from EMB than lymphocytic myocarditis. Most cases of giant cell myocarditis have an autoimmune origin, but a postviral etiology has been suggested. Targeted treatments are shown in table 4 and combination treatments in figure 1 of the supplementary data.2,32,33,57
 and necrosis with myocardial fiber disruption (arrowhead) (hematoxylin-eosin, original magnification × 200). B: lymphocytic myocarditis with positive immunohistochemical criteria; the brown inclusions correspond to CD3+ lymphocytes (arrowhead). C: dilated cardiomyopathy with extensive fibrosis (bluish-green staining) (Masson")
Endomyocardial biopsy samples from patients with suspected myocarditis. A: lymphocytic myocarditis with positive Dallas criteria; lymphocytic infiltrate (asterisk) and necrosis with myocardial fiber disruption (arrowhead) (hematoxylin-eosin, original magnification × 200). B: lymphocytic myocarditis with positive immunohistochemical criteria; the brown inclusions correspond to CD3+ lymphocytes (arrowhead). C: dilated cardiomyopathy with extensive fibrosis (bluish-green staining) (Masson's trichrome, original magnification × 100). D: cardiac sarcoidosis with granulomas (arrowhead) (hematoxylin-eosin, original magnification × 200). E: another example of cardiac sarcoidosis with granulomas (arrowhead); F: eosinophilic myocarditis (eosinophils shown byarrowhead) (hematoxylin-eosin, original magnification × 400). G: giant cell myocarditis (arrowhead) (hematoxylin-eosin, original magnification × 200). H: another example of giant cell myocarditis (yellow arrows, multinucleated giant cells). I: pembrolizumab-induced myocarditis (immune checkpoint inhibitor); lymphocytic infiltrate (arrowhead) and isolated eosinophils (asterisk) (hematoxylin-eosin, original magnification × 400).
Recommendation. When immune checkpoint inhibitor-myocarditis is suspected, immunotherapy should be discontinued and corticosteroids initiated promptly.
Myocarditis associated with immune checkpoint inhibitors appears within 6 weeks of initiation this treatment. Immune checkpoint inhibitor-myocarditis can follow a fulminant course, with mortality rates in the range of 20% to 50%. While this condition is rare, with an estimated incidence of 0.04% to 1.14%,58 it is expected to become more common with the increasing use of immunosuppressive therapy. The histological pattern is a lymphocytic infiltrate, as cytotoxicity is primarily mediated by CD8+ T cells, which also mediate viral myocarditis (figure 4). High-dose corticosteroids are the mainstay of treatment (table 4).
SarcoidosisRecommendation. Corticosteroids are the treatment of choice for sarcoidosis. Treatment response and disease course (in particular the appearance of ventricular arrhythmias) should be closely monitored to assess the need for other immunosuppressive agents or ICD placement.
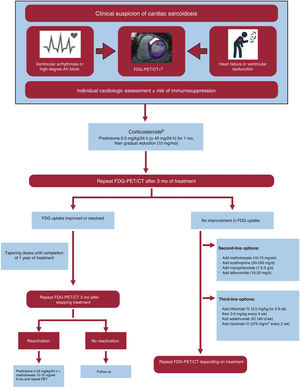
Sarcoidosis is a systemic inflammatory disease characterized by a genetic predisposition and noncaseating granulomas. It has a prevalence of 5 to 64 cases per 100 000 inhabitants and is more common among African Americans than Whites.59,60 Histological identification of noncaseating granulomas in the myocardium is a diagnostic hallmark. Sarcoidosis, however, can be diagnosed based on extracardiac histological findings combined with at least 1 of the following: suggestive findings on CMR or FDG PET, ventricular arrhythmias, advanced atrioventricular block, ventricular dysfunction, and response to immunosuppressive therapy61,62(figure 5).
![Treatment algorithm for cardiac sarcoidosis AV, atrioventricular; FDG-PET/CT, fluoro-[18F]-deoxy-2-D-glucose positron emission tomography/computed tomography; IV, intravenous; SC, subcutaneous. a Additional testing with gammagraphy or another perfusion test is advisable to help determine whether or not the inflammation is associated with a perfusion defect (more severe inflammation). Cases with perfusion defects without associated inflammation may correspond to established fibrosis. b If high-dose corticosteroids are contraindicated or poorly tolerated, a combined regimen of low-dose corticosteroids + methotrexate can be initiated (prednisone 0.25 mg/kg/24 h + methotrexate 10-15 mg/wk).](https://static.elsevier.es/multimedia/18855857/0000007700000008/v1_202407290712/S1885585724001567/v1_202407290712/en/main.assets/gr5.jpeg?xkr=eyJpdiI6ImllUG1ZWkI5U1VZUW9OWDJidU1DVkE9PSIsInZhbHVlIjoiZGtWRlZCVUd5enJkbDF4c3hxdHR6U1FtdGd2bmF4STdvMXdUQ2xNU2E2S08xZ3QzMU1vK1hTSkdRaEVJYkF0TjN5d2gxM0JKVFVzRllMSG9nQWRXN1hma3k3MlRlKzliak5pQkxCRldFNzE1U0VCRHNYeVFCNjRCbk5XbG8rY1RpRFV0ZzIyTlRBemU3VzRXVVZUU3hoUGhRL3hOQ0tkeGRLMStQeDJWQ2E2NEtUSWJ5bFdlMXYyeExJMTl1cFZHdDhiVjVYTUJDd0J4OTZxeWpvRFNOK05vc3RHMFdwdFJiZEc5cGpmU3R5SVQ5SkV5bWpRU05EZVpSUXc1aGZ1VEhYZlloaGFWY1JZam5GL014OXNsNG1tOTdybmsxZmFYb1NlS2JlK3RjV0k9IiwibWFjIjoiMDgwOGJlMDZiMDIyZjIyZWI5MmY5OWQwYjZkY2FhNjE2ZTg4YWQyMjMzMGI1MmIxMDA5NGY5ZjU4MjI4NGQ4MCIsInRhZyI6IiJ9 "Treatment algorithm for cardiac sarcoidosis AV, atrioventricular; FDG-PET/CT, fluoro-[18F]-deoxy-2-D-glucose positron emission tomography/computed tomography; IV, intravenous; SC, subcutaneous. a Additional testing with gammagraphy or another perfusion test is advisable to help determine whether or not the inflammation is associated with a perfusion defect (more severe inflammation). Cases with perfusion defects without associated inflammation may correspond to established fibrosis. b If high-dose corticosteroids are contraindicated or poorly tolerated, a combined regimen of low-dose corticosteroids + methotrexate can be initiated (prednisone 0.25 mg/kg/24 h + methotrexate 10-15 mg/wk).")
Treatment algorithm for cardiac sarcoidosis AV, atrioventricular; FDG-PET/CT, fluoro-[18F]-deoxy-2-D-glucose positron emission tomography/computed tomography; IV, intravenous; SC, subcutaneous.
a Additional testing with gammagraphy or another perfusion test is advisable to help determine whether or not the inflammation is associated with a perfusion defect (more severe inflammation). Cases with perfusion defects without associated inflammation may correspond to established fibrosis.
b If high-dose corticosteroids are contraindicated or poorly tolerated, a combined regimen of low-dose corticosteroids + methotrexate can be initiated (prednisone 0.25 mg/kg/24 h + methotrexate 10-15 mg/wk).
Treatment of cardiac sarcoidosis includes immunosuppressive therapy to help reduce the myocardial inflammation and supportive heart failure treatment to address the effects of myocardial damage and fibrosis (table 4, figure 5). European guidelines recommend primary prevention ICD in the following cases: a) need for cardiac pacing; b) extensive fibrosis on CMR after resolution of inflammation, regardless of LVEF, and c) induction of sustained ventricular tachycardia in an electrophysiological study in patients with a LVEF of 35% to 50% (class IIa recommendation).63
Eosinophilic myocarditisA definitive diagnosis of eosinophilic myocarditis requires confirmation of characteristic histological findings, namely, a diffuse inflammatory infiltrate with predominant eosinophils and extensive necrosis, and, on occasions, mural and intravascular thrombi (figure 4). Corticosteroids are the mainstay of treatment (table 4). Causative drugs or substances should be immediately withdrawn in patients with eosinophilic myocarditis caused by a hypersensitivity reaction. Imatinib is used to treat myeloproliferative variants of hypereosinophilic syndrome.64 Combination immunosuppressive therapy with corticosteroids and cyclophosphamide, azathioprine, or methotrexate is recommended for eosinophilic granulomatosis with polyangiitis.64 Finally, eosinophilic myocarditis associated with Toxocara canis infection must be treated with albendazole and corticosteroids.2
Myocarditis associated with COVID-19 and SARS-CoV-2 vaccinationRecommendation. Myocarditis associated with isolated cases of nonsevere SARS-CoV-2 infection or vaccination-induced symptoms does not require any specific treatment other than standard COVID-19 treatment or the recommendation to avoid another vaccine dose.
Myocarditis has been linked to both SARS-CoV-2 infection and vaccination, although it is more common in infections.65 Intravenous corticosteroids combined with tocilizumab or immunoglobulins are recommended for presentations with a fulminant course or associated with systemic inflammatory syndrome.66 There have been isolated reports of myocarditis/pericarditis following mRNA vaccination, mostly in young men. The cases follow a benign course and typically occur a few days after the second or third vaccine dose.65-67
Chronic inflammatory myocarditis or cardiomyopathyRecommendation. The treatment of chronic inflammatory myocarditis or cardiomyopathy should be individualized based on EMB findings and the identification of specific viruses or specific systemic inflammatory/autoimmune causes.
Chronic inflammatory cardiomyopathy can occur in association with systemic autoimmune diseases such as lupus and systemic sclerosis or inflammatory diseases affecting specific organs or systems (eg, intestinal and herpes-related inflammatory diseases). There may not, however, always be an underlying systemic disorder. Treatment should be decided on a case-by-case basis by multidisciplinary teams and target the systemic disease. More aggressive treatments are recommended, as chronic inflammatory cardiomyopathy has a worse prognosis.68 Treatment options, which vary according to EMB findings (positive vs negative findings for inflammation and viral genomes), are shown in figure 3.
Immunosuppressive therapy with prednisone and azathioprine has been associated with improved ventricular function in EMB-confirmed inflammation without a viral agent.30 Immunosuppressive agents can also be used in patients with latent parvovirus B19 or human herpesvirus (HHV) 6 infection (without active replication, EMB-proven inflammation, and low copy numbers of viral DNA).69
Myocarditis in pediatric patientsMyocarditis has 2 notable peaks in pediatric populations: one during early childhood (associated with a worse prognosis) and another during adolescence. Up to 5% of children on pediatric heart transplant wait lists have myocarditis, illustrating the potential severity of this condition. Young age has been significantly correlated with worse clinical outcomes, particularly in neonates.70
Myocarditis in pediatric patients has a range of causes, including autoimmune diseases, infections, hypersensitivity, and toxicity. Viral agents are the most common cause. Notably, the most frequently detected pathogen in EMB studies prior to the COVID-19 era was parvovirus B19 (59%), followed by HHV-6, enterovirus, cytomegalovirus, Epstein-Barr virus, influenza A and B viruses, and coronavirus.71
EMB has generally been considered the technique of choice for identifying cardiotropic viruses and guiding treatment and prognosis. Its limited sensitivity, however, is compounded by a high risk of complications, particularly in neonates and infants. EMB should thus only be used in centers with demonstrated expertise and when it is expected to influence clinical management. ECG techniques can be used to optimize sampling points. CMR, reinforced by the Lake Louise criteria, is gradually replacing EMB. Because of its limited sensitivity in pediatric patients, however, CMR should only be performed in acute stages. Between 11% and 55% of patients with pediatric myocarditis may require mechanical circulatory support, either as a bridge to recovery or in preparation for transplantation.70
Additional information on the treatment of specific forms of myocarditis can be found in the supplementary data.
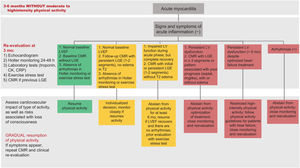
RISK STRATIFICATION, LONG-TERM FOLLOW-UP, AND SPORTRecommendation. A diagnosis should always be followed by risk stratification and a structured follow-up plan.
Patients with myocarditis can be categorized into 1 of 3 risk groups according to clinical course and diagnostic test results. The follow-up protocol in each case varies (table 5). Both European72 and US guidelines73 recommend that patients with myocarditis avoid moderate to high-intensity exercise for 3 to 6 months to allow time for the inflammation to resolve. Time to resumption of activity should be decided on a case-by-case basis depending on the patient's overall evaluation and level of risk (figure 6).
Follow-up of patients with myocarditis according to risk profile
| Low risk |
| Stable hemodynamicsNonspecific changes on ECG (ST, T wave)Absence of complex rhythmsPreserved LVEF, no alterations in regional contractilityCMR showing edema, minimal or no LGE |
| Follow-upClinical evaluation in 3-6 mo with ECG and laboratory tests (complete blood count, biochemistry including hepatic and renal profile, acute phase reactants, and troponin)If normal, discharge to primary careIf ECG changes and/or laboratory alterations, switch to intermediate-risk follow-up protocol |
| Intermediate risk |
| Stable hemodynamicsNonspecific ECG changes (ST, T wave)Absence of complex rhythmsLVEF 35%-50% with or without alterations in regional contractilityLGE on CMR (above all in ventricular septum) |
| Follow-upClinical evaluation at 3, 6, and 12 mo with ECG and laboratory tests (complete blood count, biochemistry including hepatic and renal profile, acute phase reactants, and troponin); discharge or annual follow-up according to CMR results at 12 moFollow-up echocardiogram at 6 moCMR at 12 moExercise stress test before resuming sport |
| High risk |
| Unstable hemodynamicsHeart failureComplex arrhythmias (severe conduction disturbance or sustained ventricular arrhythmias)Severely depressed LVEF (<35%)Extensive fibrosis on CMR, with involvement of ≥ 3 segments or a pattern indicative of poor prognosis (septal, ring-like) |
| Follow-upClinical evaluation at 3, 6, and 12 mo with ECG and laboratory tests (complete blood count, biochemistry including hepatic and renal profile, acute phase reactants, and troponin)Echocardiogram and Holter ECG 3 months after diagnosis, then every 3-6 mo or annually depending on resultsEndomyocardial biopsy at 3 mo if uncertain cause and no response to optimized medical treatmentCMR at 6 moExercise stress test before resuming sport |
CMR, cardiac magnetic resonance imaging; ECG, electrocardiogram; LGE; late gadolinium enhancement; LVEF, late ventricular ejection fraction.

No specific funding was received for this consensus statement.
USE OF ARTIFICIAL INTELLIGENCENo artificial intelligence tools were used.
AUTHORS’ CONTRIBUTIONSAll the authors contributed to the literature search and drafting of this statement (F. Domínguez, A. Uribarri, J.M. Larrañaga-Moreira, L. Guerrero-Ruiz, P. Pastor-Pueyo, J. Gayán-Ordás, B. Fernández-González, A. Esteban-Fernández, M. Barreiro, S. López Fernández, F. Gutiérrez-Larraya Aguado, and D. Pascual Figal). F. Domínguez and D. Pascual Figal designed the document and served as coordinators for the project.
CONFLICTS OF INTERESTThere are no conflicts of interest.