A known long QT syndrome-related mutation in Nav1.5 cardiac channels (p.R1644H) was found in 4 members of a Spanish family but only 1 of them showed prolongation of the QT interval. In the other 3 relatives, a novel missense mutation in Cav1.2 cardiac channels was found (p.S1961N). Here, we functionally analyzed p.S1961N Cav1.2 channels to elucidate whether this mutation regulates the expressivity of the long QT syndrome phenotype in this family.
MethodsL-type calcium current (ICaL) recordings were performed by using the whole-cell patch-clamp technique in Chinese hamster ovary cells transiently transfected with native and/or p.S1961N Cav1.2 channels.
ResultsExpression of p.S1961N channels significantly decreased ICaL density. Using Ba as a charge carrier to suppress the Ca-dependent inactivation of Cav1.2 channels, we demonstrated that the mutation significantly accelerates the voltage-dependent inactivation of Cav1.2 channels decreasing the inactivation time constant. As a consequence, the total charge flowing through p.S1961N Cav1.2 channels significantly decreased. The effects of the p.S1961N Cav1.2 and p.R1644H Nav1.5 mutations alone or their combination on the action potential (AP) morphology were simulated using a validated model of the human ventricular AP. The p.S1961N Cav1.2 mutation shortens the AP duration and abrogates the prolongation induced by p.R1644H Nav1.5 channels.
ConclusionsThe p.S1961N mutation in Cav1.2 channels decreased the ICaL, an effect which might shorten ventricular AP. The presence of the loss-of-function Cav1.2 mutation could functionally compensate the prolonging effects produced by the Nav1.5 gain-of-function mutation.
Keywords
The long QT syndrome (LQTS) is an inherited primary arrhythmogenic syndrome characterized by a prolonged QT interval on the electrocardiogram (ECG) due to delayed ventricular repolarization and is associated with syncope, seizures, ventricular tachycardia (mainly torsade de pointes), and a high risk of sudden cardiac death.1,2 Ventricular action potential (AP) duration (APD) is the result of a balance between depolarizing (Na and Ca) and repolarizing (K) currents. Thus, both an increase in the depolarizing or a decrease in the repolarizing currents could give rise to the different types of LQTS.1,2
LQTS type 3 is due to gain-of-function mutations in the SCN5A gene, which encodes the α-subunit of the sodium channels (Nav1.5) responsible for the cardiac sodium current (INa).3 These mutations usually modify Nav1.5 channel kinetics by several biophysical mechanisms ultimately leading to an increase in the late component of the INa (INaL) that allows a persistent inward current during the plateau phase of the ventricular AP that lengthens the APD.3
CACNA1C, CACNB2, and CACNA2D1 genes encode the pore forming α-subunit (Cav1.2) and the β2 and α2δ ancillary subunits, respectively, that form the cardiac Ca channel which generates the L-type Ca current (ICaL).4 Loss-of-function mutations in CACNA1C and CACNB2 genes shorten the APD and have been associated with Brugada syndrome and short QT syndrome.5,6 Conversely, gain-of-function mutations in the CACNA1C gene lead to prolongation of the ventricular APD and LQTS type 8.7
In this study, we identified a previously characterized gain-of-function mutation in SCN5A (encoding p.R1644H Nav1.5)8 in 4 relatives of a Spanish family in which only 1 of the carriers had a LQTS type 3 phenotype. Genetic analysis of the other 3 relatives who had normal corrected QT (QTc) values demonstrated that they also carry a novel missense mutation in the CACNA1C gene encoding for p.S1961N Cav1.2 channels. Therefore, these family members harbour 2 variants in different genes (digenic heterozygosity). Thus, this study aimed to characterize the electrophysiological properties of p.S1961N Cav1.2 channels to elucidate whether the presence of this variant can ameliorate the APD prolongation and LQTS produced by the p.R1644H Nav1.5 mutant.
METHODSGenetic AnalysisClinical evaluation, including 12-lead ECG, of the family members was performed at the Hospital Universitario Virgen de las Nieves (Granada, Spain). The study was approved by the local ethics committee and conforms to the principles outlined in the Declaration of Helsinki. Each participant gave written informed consent.9–11
The whole codifying sequence and the flanking intronic regions of KCNQ1, KCNH2, SCN5A, KCNJ2, KCNJ8, CACNA1C, AKAP9, KCNE1, and KCNE2 genes were sequenced using an Illumina 1500 Hiseq next-generation sequencing platform. Potentially pathogenic variants were confirmed using Sanger sequencing.9–11 The pathogenicity of the variants was predicted according to the current recommendations of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (see details in the “Genetic analysis” section of the supplementary material).12 Other exonic nonsynonymous variants found, not predicted as pathogenic, are described in the .
Mutagenesis and Cell TransfectionThe p.S1961N substitution in Cav1.2 (NP_000710.5) was introduced using the QuikChange Site-Directed Mutagenesis kit (Stratagene, United States) and confirmed by direct deoxyribonucleic acid (DNA) sequencing.5,9,11,13 Chinese hamster ovary cells were transiently transfected with the cDNA encoding native (WT) or mutated Cav1.2 channels and the α2δ and β subunits (1:1.7:4 ratio).
Patch-clampingCurrents were recorded using the whole-cell patch-clamp configuration following previously described methods.9,13 Uncompensated access resistance, cell capacitance, and peak ICaL amplitude generated by WT Cav1.2 channels were 1.8 ± 0.4 M?, 16.2 ± 1.2 pF, and -521 ± 60 pA (n = 25), respectively. Therefore, no significant voltage errors (< 5mV) due to series resistance were expected with the micropipettes used.
Mathematical Modelling of Ventricular Action PotentialTo simulate the shapes of human ventricular AP, we employed the O’Hara-Rudy model previously validated and used for similar purposes.14 The model was run at different driving frequencies (0.1-3Hz) under baseline conditions (WT) or by incorporating the changes in INaL and ICaL produced by p.R1644H Nav1.5 and p.S1961N Cav1.2 mutations, respectively.
Statistical AnalysisResults are expressed as mean ± standard error of the mean. Paired or unpaired t test or 1-way ANOVA followed by the Newman-Keuls test were used to assess statistical significance where appropriate. To take into account repeated sample assessments, data were analyzed with multilevel mixed-effects models. A value of P < .05 was considered statistically significant. An expanded Methods section is available in the supplementary material.
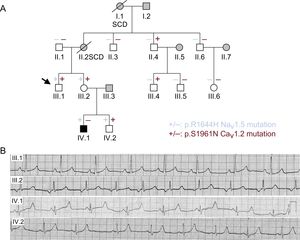
RESULTSCase Description and Genetic AnalysisThe proband (III.1) (arrow in Figure 1A) had a syncopal episode when he was 28 years old after a single dose of ciprofloxacin, a QT prolonging drug.15 This suggested that he might have a genetic predisposition to drug-induced QT prolongation. Conversely, his clinical evaluation revealed a normal ECG with a Bazzett QTc of 420ms (Figure 1B). However, both his mother (II.2) and grandmother (I.1), who had never experienced syncopal episodes, suddenly died at rest when they were 49 years old (clinical or genetic data were not available). Therefore, his family history of sudden cardiac death encouraged the evaluation of the proband's relatives. The proband's sister (III.2) showed an inferior atrial rhythm at 60 beats per minute, normal PR and QRS durations, and a slightly widened T wave (Figure 1A,1B). However, her QT properly shortened during exercise and the QTc was also normal (430ms). One of the proband's nephews (IV.1), a 13-year-old boy, had an ECG with a late T wave after a long flat ST-segment, which resulted in a markedly prolonged QTc (558ms) (Figure 1B). Conversely, IV.2 showed a QTc of 420ms with normal T wave (Figure 1B). The rest of the relatives studied (white symbols in Figure 1A) have been and are currently asymptomatic, with completely normal ECGs.
![A: pedigree of the family (females [○] and males [□]). The arrow indicates the proband. Diagonal lines indicate deceased patients. + and – indicate individuals with and without the p.R1644H Nav1.5 (blue) and p.S1961N Cav1.2 (red) variants, respectively. Black symbols indicate long QT syndrome-affected individuals, while grey symbols depict individuals without genetic testing. White symbols represent studied individuals not affected by any clinical phenotype. B: electrocardiogram of III.1, III.2, IV.1, and IV.2. SCD, sudden cardiac death.](https://static.elsevier.es/multimedia/18855857/0000007200000004/v1_201903190629/S1885585718300987/v1_201903190629/en/main.assets/gr1.jpeg?xkr=eyJpdiI6IkNpZHRweVg3V09NekZaYm96R3JsNHc9PSIsInZhbHVlIjoiRVZEMUNaMkFhemxnSzcxandvdktSaUNUeTNNZ3VMeTRCUVN0OUNGMVlNcVlDNmg2aHR6NWUvSTBvdGJ6VHE1VEc5eENxeTliL0x0S2w0TVEwKzNhaXBza3ZjZkYxTjdRcFlzb3R0YmcxMzNNR3YwT2tGOWRrdDk4RTRGSGZ0cFB0aVlyM2JDSGtDTjNIOVdrNFhBVW9iTDMyTTVpSGE5a2F4bE8wYmszR1BOV1R1QTc3bkJKUmVkOWptYkQzV1djaEM5bkdLbVF1M3dZRVBMZko5VlJ4MVljTHpoZm1aOTZWYXptdG1Xd2s2Yy9BWnBnQmlJTkhCUmlleEMzckxscXRldktMY0xuOW5IUndqcFJYUU4xT3dDeXc3bkc0MDVOWHdvYlNhOEMxNXc9IiwibWFjIjoiYjljYzg0NzZhMzg1OGYyZDQxMTgzZDNmODZmY2EzMzZiZmJhMzY2ZTY5MjMxZDk2Yjk4YjVhNTZhZTUxMzdkNSIsInRhZyI6IiJ9 "A: pedigree of the family (females [○] and males [□]). The arrow indicates the proband. Diagonal lines indicate deceased patients. + and – indicate individuals with and without the p.R1644H Nav1.5 (blue) and p.S1961N Cav1.2 (red) variants, respectively. Black symbols indicate long QT syndrome-affected individuals, while grey symbols depict individuals without genetic testing. White symbols represent studied individuals not affected by any clinical phenotype. B: electrocardiogram of III.1, III.2, IV.1, and IV.2. SCD, sudden cardiac death.")
A: pedigree of the family (females [○] and males [□]). The arrow indicates the proband. Diagonal lines indicate deceased patients. + and – indicate individuals with and without the p.R1644H Nav1.5 (blue) and p.S1961N Cav1.2 (red) variants, respectively. Black symbols indicate long QT syndrome-affected individuals, while grey symbols depict individuals without genetic testing. White symbols represent studied individuals not affected by any clinical phenotype. B: electrocardiogram of III.1, III.2, IV.1, and IV.2. SCD, sudden cardiac death.
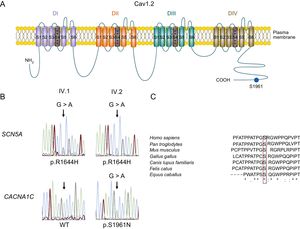
Considering the clinical data, a genetic analysis by means of next-generation sequencing (see Methods) was offered to both the proband, who initially refused it, and his sister, who accepted. Genotyping of the proband's sister identified 2 mutations (digenic heterozigosity). These mutations were thereafter sequenced, using the Sanger method, in the proband (who finally accepted) and the rest of the relatives studied. The first mutation, predicted as pathogenic (see the “Genetic analysis” section of the supplementary material), appeared in SCN5A (NC_000003.11:g.38592932C>T according to GRCh37) leading to the arginine-to-histidine substitution of the residue 1644 (p.R1644H) in Nav1.5 channels. This mutation was previously described in several families with LQTS type 3 and has never been related to any other arrhythmic phenotype, including the Brugada syndrome.8,16–18 Functional studies revealed that the p.R1644H mutation produces a marked increase in INaL (gain-of-function), which explains the APD and QT prolongation.8,16 The p.R1644H Nav1.5 mutation was also found in the proband and nephews, but not in any other of the relatives tested (Figure 1A). In the proband's sister, the proband, and 1 of his nephews (Figure 1A) a mutation in the CACNA1C gene was also identified (NC_000012.11:g.2797710G>A) (Figure 2B) that leads to a serine-to-asparagine substitution in the Cav1.2 channel at position 1961 (p.S1961N) (Figure 2A). The serine residue is highly preserved at the equivalent position among different species (Figure 2C) and the mutation has not been annotated previously in any public database. Importantly, the p.S1961N Cav1.2 mutation is also carried by II.4 and III.4 but both individuals had normal ECGs and have never experienced any arrhythmic event.
 of the SCN5A gene leading to the missense mutation p.R1644H in both individuals (top) and a WT sequence (left) and the heterozygous mutation (NC_000012.11:g.2797710G>A) (right) of the CACNA1C gene, which results in the p.S1961N missense mutation (bottom). C: sequence alignment of the regions surrounding S1961 residue in Cav1.2 channels in several species. The box highlights the conservation of this residue. “*” identical residues. “:” and “.” indicate conservation between groups of strongly and weakly similar properties, respectively. DNA, deoxyribonucleic acid; WT, native form of the channel.")
A: scheme of the L-type cardiac calcium channel. B: DNA sequence chromatograms of IV.1 and IV.2 depicting the heterozygous variation (NC_000003.11:g.38592932C>T) of the SCN5A gene leading to the missense mutation p.R1644H in both individuals (top) and a WT sequence (left) and the heterozygous mutation (NC_000012.11:g.2797710G>A) (right) of the CACNA1C gene, which results in the p.S1961N missense mutation (bottom). C: sequence alignment of the regions surrounding S1961 residue in Cav1.2 channels in several species. The box highlights the conservation of this residue. “*” identical residues. “:” and “.” indicate conservation between groups of strongly and weakly similar properties, respectively. DNA, deoxyribonucleic acid; WT, native form of the channel.
Therefore, family members who showed digenic heterozygosity do not exhibit LQTS type 3 phenotype, while IV.1, who is the only one who carries only the SCN5A mutation, does. Thus, we decided to functionally characterize the p.S1961N Cav1.2 mutation to elucidate whether the presence of this variant can ameliorate the APD prolongation and LQTS type 3 produced by the p.R1644H Nav1.5 mutant.
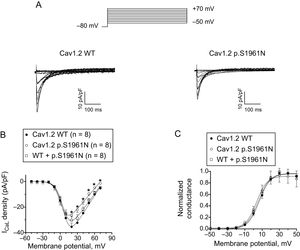
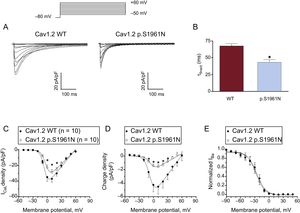
Functional Characterization of p.S1961N Cav1.2 ChannelsFigure 3A shows ICaL traces recorded at potentials between -50 and +70mV in Chinese hamster ovary cells expressing human cardiac WT or p.S1961N Cav1.2 channels along with the α2δ and β subunits. Peak density of the currents generated by p.S1961N Cav1.2 channels (-26.4 ± 1.4 pA/pF; P < .05) was approximately 25% lower than that generated by WT channels (-35.5 ± 3.0 pA/pF) (Figure 3A, 3B). In another group of experiments, cells were cotransfected (0.5:0.5 ratio) with the cDNA encoding WT and p.S1961N Cav1.2 channels considering the heterozygous condition of the mutation carriers. Under these conditions, peak current density was not statistically different to that generated by WT and p.S1961N channels separately (Figure 3B; P > .05). Figure 3C shows the voltage dependence of the activation of WT, p.S1961N, and WT+p.S1961N Cav1.2 channels obtained by plotting the normalized conductance (estimated by using equation #1; see section patch-clamping of the supplementary material) as a function of the membrane potential. The fit of a Boltzmann function to the data, yielded the midpoint (Vh) and the slope (k) of the curves, which almost overlap (Figure 3C). Thus, Vh and k values of the 3 curves were not significantly different (Table). The fit of the equation #2 (see section patch-clamping of the supplementary material) to the current density-voltage data in each experiment yielded the Erev which averaged 74.9 ± 2.7mV for WT channels. The mutation, either expressed alone or in combination with WT channels, did not modify the Erev (P > .05) (Table).
 and conductance-voltage curves (C) for ICaL generated by WT, p.S1961N or WT+p.S1961N Cav1.2 channels. Each point represents the mean ± standard error of the mean of (n) experiments. In B, *P < .05 vs Cav1.2 WT. ICaL, L-type Ca current. WT, native form of the channel.")
A: currents generated by WT or p.S1961N Cav1.2 channels. B,C: mean density-voltage (B) and conductance-voltage curves (C) for ICaL generated by WT, p.S1961N or WT+p.S1961N Cav1.2 channels. Each point represents the mean ± standard error of the mean of (n) experiments. In B, *P < .05 vs Cav1.2 WT. ICaL, L-type Ca current. WT, native form of the channel.
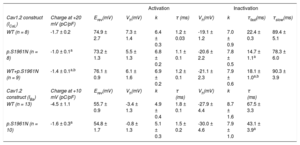
Biophysical Properties of WT and Mutant Cav1.2 Channels
| Activation | Inactivation | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cav1.2 construct (ICaL) | Charge at +20 mV (pC/pF) | Erev(mV) | Vh(mV) | k | τ (ms) | Vh(mV) | k | τfast(ms) | τslow(ms) |
| WT (n = 8) | -1.7 ± 0.2 | 74.9 ± 2.7 | 7.3 ± 1.4 | 6.4 ± 0.3 | 1.2 ± 0.03 | -19.1 ± 1.2 | 7.0 ± 0.9 | 22.4 ± 0.3 | 89.4 ± 5.1 |
| p.S1961N (n = 8) | -1.0 ± 0.1a | 73.2 ± 1.3 | 5.5 ± 1.3 | 6.8 ± 0.2 | 1.1 ± 0.1 | -20.6 ± 2.2 | 7.8 ± 0.5 | 14.7 ± 1.1a | 78.3 ± 6.0 |
| WT+p.S1961N (n = 9) | -1.4 ± 0.1a,b | 76.1 ± 0.9 | 6.1 ± 1.6 | 6.9 ± 0.2 | 1.2 ± 0.1 | -21.1 ± 2.3 | 7.9 ± 0.6 | 18.1 ± 1.0a,b | 90.3 ± 3.9 |
| Cav1.2 construct (IBa) | Charge at +10 mV (pC/pF) | Erev(mV) | Vh(mV) | k | τ (ms) | Vh(mV) | k | τ (ms) | |
| WT (n = 13) | -4.5 ± 1.1 | 55.7 ± 0.9 | -3.4 ± 1.3 | 4.9 ± 0.4 | 1.8 ± 0.1 | -27.9 ± 4.4 | 8.7 ± 1.6 | 67.5 ± 3.3 | |
| p.S1961N (n = 10) | -1.6 ± 0.3a | 54.8 ± 1.7 | -0.8 ± 1.3 | 5.1 ± 0.3 | 1.5 ± 0.2 | -30.0 ± 4.6 | 7.9 ± 1.0 | 43.1 ± 3.9a | |
Erev, reversal potential; IBa, current generated using Ba as a charge carrier; ICaL: L-type Ca current; τ, time constants of activation (ICaL)/inactivation (IBa) obtained by fitting a monoexponential function to the current traces recorded at +20mV. τfast and τslow, fast and slow time constants of inactivation (ICaL) obtained by fitting a biexponential function to the current traces recorded at +20mV; Vh and k, midpoint and slope values of the activation and inactivation curves; WT, native form of the channels.
Values are the mean ± standard error of the mean of (n) experiments in each group.
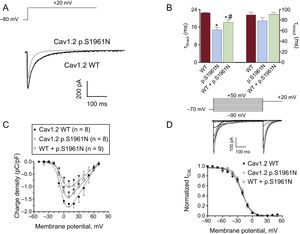
Figure 4A shows superimposed ICaL traces elicited when applying 500 ms-pulses from -80 to +20mV in cells expressing WT or p.S1961N Cav1.2 channels (currents generated by WT+p.S1961N channels were omitted for clarity). Activation kinetics of Cav1.2 channels was not modified by the p.S1961N mutation alone or cotransfected with WT channels (Table; P > .05). Conversely, the mutation significantly accelerated the time course of current inactivation (Figure 4A), decreasing the fast time constant of inactivation (τfast) calculated by the biexponential fit to the current decline (Figure 4B and Table). Interestingly, τfast of the current generated by WT+p.S1961N channels was intermediate (Figure 4B and Table). Therefore, to accurately analyze the effects produced by the p.S1961N mutation on the total Ca influx we measured the charge density generated by WT, p.S1961N, and WT+p.S1961N Cav1.2 channels at each membrane potential (Figure 4C). The density was calculated by normalizing the charge crossing the membrane, estimated from the integral of current traces, to the cell capacitance (methods of the supplementary material). Due to the combined effects of the mutation on the peak current density and on the inactivation kinetics, the charge was significantly reduced by the p.S1961N mutation either alone or together with WT (Figure 4C).

A: superimposed current traces generated at +20mV by WT or p.S1961N Cav1.2 channels. B: fast and slow time constants of inactivation obtained at +20mV in cells expressing WT, p.S1961N or WT+p.S1961N Cav1.2 channels. C: charge estimated as the integral of the current traces recorded as a function of the membrane potential in the 3 groups. D: Top: currents generated by WT Cav1.2 channels using the protocol shown. Bottom: inactivation curves for WT, p.S1961N or WT+p.S1961N Cav1.2 channels. Each bar/point represents the mean ± standard error of the mean of ≥ 8 experiments. *P < .05 vs Cav1.2 WT. WT, native form of the channel.
Figure 4D (top) shows ICaL traces generated by WT Cav1.2 channels with the protocol used to assess the voltage dependence of inactivation and construct the inactivation curves (methods of the supplementary material) (Figure 4D bottom). Neither the Vh nor the k of the inactivation curves were modified by the p.S1961N mutation either alone or in combination with WT channels (Figure 4D and Table).
The p.S1961N Mutation Accelerated the Cav1.2 Voltage-dependent InactivationInactivation of Cav1.2 channels consists of 2 independent mechanisms, the Ca-dependent (CDI) and the voltage-dependent inactivation (VDI). We tested the effects of p.S1961N mutation on the VDI by using Ba as charge carrier since Ba currents (IBa) do not exhibit CDI.19Figure 5A shows IBa traces generated in 2 Chinese hamster ovary cells transfected with WT or p.S1961N Cav1.2 channels, respectively. Inactivation decay of IBa defined by a monoexponential function was markedly accelerated by the mutation (Figure 5B and Table). Furthermore, the mutation significantly decreased the peak IBa and Ba charge densities, the decrease in the charge being greater than that of the peak current (Figure 5C and Figure 5D). Finally, the voltage dependence of IBa inactivation was not modified by the mutation (Figure 5E and Table). All these results strongly suggested that p.S1961N mutation accelerates the VDI of Cav1.2 channels.
, mean current density- (C), charge-density voltage (D), and inactivation (E) curves for IBa generated by WT or p.S1961N Cav1.2 channels. Each point represents the mean ± standard error of the mean of 10 experiments. In B-D, *P < .05 vs Cav1.2 WT. WT, native form of the channel.")
A: IBa traces generated by WT or p.S1961N Cav1.2 channels. B-E: inactivation time constants obtained at +10mV (B), mean current density- (C), charge-density voltage (D), and inactivation (E) curves for IBa generated by WT or p.S1961N Cav1.2 channels. Each point represents the mean ± standard error of the mean of 10 experiments. In B-D, *P < .05 vs Cav1.2 WT. WT, native form of the channel.
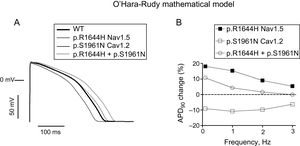
The effects of the p.S1961N Cav1.2 and p.R1644H Nav1.5 mutations, alone or combined, on the AP characteristics were simulated using a validated model of human endocardial ventricular myocytes (Figure 6).14 The model was run in baseline conditions (WT) or by introducing the kinetic and conductance changes produced by the p.S1961N Cav1.2 mutation. To simulate the effects of the p.R1644H mutation the INaL conductance was increased ≈3-fold, as previously described.8Figure 6A shows the simulated AP traces obtained at 1Hz. p.R1644H Nav1.5 lengthened the APD20, APD50, and APD90 by 17.4%, 17.1%, and 15.2%, respectively. Due to the ICaL reduction, p.S1961N Cav1.2 shortened APD20, APD50, and APD90 by 21.7%, 14.3%, and 10.8%, respectively (Figure 6A). Interestingly, the AP traces corresponding to p.R1644H+p.S1961N and WT were almost undistinguishable, and only a small prolongation of the APD90 (4.3%) was observed. Figure 6B depicts the APD90 change (prolongation or shortening) induced by each mutation in cells driven at different frequencies (0.1-3Hz). Prolongation induced by p.R1644H Nav1.5 was maximum at low frequencies and decreased progressively at faster rates. The shortening induced by p.S1961N Cav1.2 was similar at frequencies between 0.1 and 2Hz (≈10%), decreasing slightly at 3Hz. In the presence of p.S1961N, the prolongation induced by p.R1644H was reduced, being completely suppressed at high frequencies (2 and 3Hz).
, or in the presence of the p.R1644H Nav1.5 and p.S1961N Cav1.2 mutations alone or in combination. B: percentage of APD90 prolongation (positive values) or shortening (negative values) induced by each mutation alone or in combination in endocardial cells driven at 0.1-3Hz. APD, action potential duration; WT, native form of the channel.")
A: action potential traces generated by the O’Hara-Rudy model in endocardial ventricular myocytes at 1Hz in baseline conditions (WT), or in the presence of the p.R1644H Nav1.5 and p.S1961N Cav1.2 mutations alone or in combination. B: percentage of APD90 prolongation (positive values) or shortening (negative values) induced by each mutation alone or in combination in endocardial cells driven at 0.1-3Hz. APD, action potential duration; WT, native form of the channel.
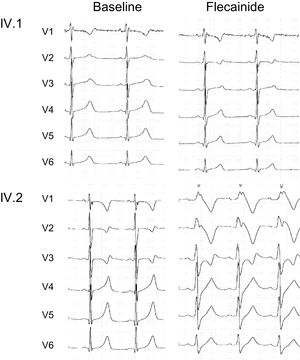
Expert consensus recommendations on LQTS therapeutic interventions consider that INaL inhibitors, such as flecainide, can be useful as add-on therapy for the treatment of LQTS type 3 patients.20 Accordingly, we tested the safety and efficacy of flecainide by infusing the drug (2mg/kg) under a controlled situation, before the implementation of a treatment of the IV.1 patient with this drug. Figure 7A shows that in the gain-of-function SCN5A mutant carrier, flecainide shortened the QT interval (QTc decreased from 558 to 470ms) without inducing any arrhythmic response. Therefore, in this child, oral flecainide was added-on the maximal tolerated dose of atenolol (50mg od) for arrhythmic event prophylaxis.
 of IV.1 (top) and IV.2 (bottom) before (baseline) and after 10minutes of intravenous infusion of flecainide at 2mg/kg.")
Conversely, loss-of-function CACNA1C mutations have been related to Brugada syndrome5 which is definitively diagnosed when a type I ST-segment elevation in precordial leads is observed after intravenous administration of flecainide. Since our functional analysis demonstrated that the p.S1961N Cav1.2 mutation reduces the ICaL density, we wondered whether the presence of the CACNA1C mutation would lead to a Brugada syndrome-like pattern in the presence of flecainide.11 Therefore, we performed a flecainide test in IV.2, who carries both the SCN5A and the loss-of-function CACNA1C mutation. Interestingly, infusion for 10minutes with flecainide (also 2mg/kg) in the patient IV.2 produced a QRS widening and a Brugada syndrome-like pattern, without modifying the QTc (Figure 7).
DISCUSSIONHere we functionally describe a novel missense mutation located at the distal C-terminal domain of Cav1.2 (p.S1961N). This mutation was found in a proband and 4 of his relatives belonging to a Spanish family with history of syncope and sudden cardiac death. The results demonstrated that the p.S1961N mutation decreased the ICaL density, which probably shortens the duration of the plateau phase of the cardiac AP.
The oldest nephew of the proband had LQTS type 3 and was found to carry the p.R1644H Nav1.5 mutation. However, his mother, his brother, and his uncle (the proband) do carry the p.R1644H Nav1.5 mutation but did not show LQTS features. This could be explained by the frequent variable LQTS expressivity among carriers of a mutation, which, in some families can be explained by the presence of multiple mutations in the same or different genes (compound or digenic heterozygosity, respectively) or by single nucleotide polymorphisms (SNPs).9,10,21 Indeed, the functional compensation between mutations or polymorphisms within the SCN5A gene has been previously described.10,22,23 Such is the case of the common Nav1.5 p.H558R polymorphism when it is presented in the same (cis) or in a different allele (trans) from the mutation.10,21,23 Furthermore, it has been shown that 2 SCN5A mutations in trans (compound heterozygosity) can interact, ameliorating their deleterious effects reciprocally.10,24 However, no additional polymorphisms or mutations were found in the SCN5A gene in any of the family members studied here.
Some SCN5A mutations can give rise to a wide spectrum of disease phenotypes among the different carriers, thus producing “overlapping syndromes”.11,25 In fact, the substitution of Arg by Cys at the same position (p.R1644C) of the Nav1.5 channels has been identified in families with either LQTS type 326 or Brugada syndrome.27 However, this is not the case in our family, in which besides the LQTS type 3 phenotype of patient IV.1, no other electrocardiographic features suggesting other arrhythmic phenotype were identified. This is consistent with data previously reported regarding other families carrying the p.R1644H gain-of-function mutation, which has exclusively been associated with LQTS type 3.17,18
p.S1961N Cav1.2 Mutation Reduces L-type Ca Current DensityAt physiologically relevant membrane potentials, the peak ICaL density generated by p.S1961N Cav1.2 channels decreased by ∼25% and the total charge crossing the membrane by ∼40%. Conversely, when cells were simultaneously transfected with WT and p.S1961N Cav1.2 channels (0.5:0.5 ratio) to “reproduce” the heterozygous condition of the participants, the current decrease was lessened. These results agree with the mild-normal phenotype of the p.S1961N mutation carriers who exhibited normal ECG and QT intervals, either when they had it concurrently with the Nav1.5 mutation (III.1, III.2, IV.2) or when not (II.4 and III.4). Importantly, the hallmark of the mutation was the acceleration of the time course of Cav1.2 channel inactivation without any alteration of the voltage dependence of either activation or inactivation.
As mentioned, inactivation of Cav1.2 channels consists of CDI and VDI components.19,28 CDI is mainly controlled by Ca/calmodulin binding to an IQ motif and is greatly inhibited when Ba is used as charge carrier.19 Molecular determinants of VDI, which include, among others, the C-terminal domain of Cav1.2 channels, are multiple and complex.28 Previous results demonstrated that mutations in the calcineurin (CaN)-binding site at the Cav1.2 channel (residues 1913-1941; NP_000710.5) markedly increase the VDI rate.29 Furthermore, it was suggested that the CaN-binding site is the VDI regulatory motif of the Ca cardiac channel,30 which partially overlaps with the protein phosphatase 2A (PP2A)-binding site (residues 1928-1970).31 According to this, the p.S1961 residue lies in the C-terminal region of the PP2A-binding site. Our results demonstrated that the p.S1961N mutation mainly increases the VDI rate of Cav1.2 channels, and thus, it could be speculated that the mutation enhances the PP2A binding and/or function. Indeed, some studies have suggested that PP2A-mediated dephosphorylation of Cav1.2 channels inhibits their function by antagonizing the cAMP-dependent protein kinase-mediated ICaL increase.31 Finally, the novel loss-of-function mutation in CACNA1C gene that we have described might, by removing the serine at position 1961, eliminate a putative phosphorylation site. Indeed, this serine is predicted as a putative phosphorylation site for phosphoinositide-dependent kinase 1 (PDPK1) by bioinformatic predictions (Group-based prediction System 3.0), although it has not been demonstrated experimentally. Therefore, further studies are needed to determine the actual mechanism by which the mutation causes an acceleration of the VDI of Cav1.2 channels.
Clinical Implications of the Functional Findings in this FamilyThe results of the mathematical model confirmed that the INaL increase produced by the p.R1644H Nav1.5 mutation could be responsible for the prolongation of the APD and QT interval duration observed in IV.1. Conversely, the slight decrease in the Ca influx during the plateau phase of the human cardiac AP produced by the p.S1961N Cav1.2 mutation could compensate the increase in the Na influx produced by the p.R1644H Nav1.5 mutation. Consequently, those family members that harbour both mutations would exhibit “almost normal” ventricular APs and ECG. However, we must keep in mind that the proband, who carries both mutations, had a syncopal episode when he was under treatment with ciprofloxacin, an antibiotic that increases the QT interval.15 Furthermore, both the mother and the grandmother of the proband, who seem to be “obligate carriers” of both mutations (the proband's father does not have any of the mutations), experienced sudden cardiac death. Of note, in patient IV.2, who also carries both mutations, flecainide infusion produced a noticeable QRS widening and a Brugada syndrome-like pattern, without any modification of the QT duration. Therefore, it can be hypothesized that the “functional compensation” between the gain-of-function and loss-of-function mutations of the Na and Ca channels, respectively, seems to be limited to conditions in which the genetic background of the patients does not concur with other factors that either prolong the ventricular repolarization (such as drugs, bradycardia, and electrolyte disturbances) or decrease cardiac excitability and/or shorten the duration of the plateau phase of the ventricular AP (such as flecainide). Therefore, it seems that in the patients carrying both mutations, either torsades de pointes- or Brugada syndrome-associated arrhythmias could be generated as a function of different proarrhythmic factors.
CONCLUSIONSThese results further support the contention that variable penetrance and phenotypic expressivity of arrhythmogenic inherited syndromes are partially attributable to genetic factors. In this context, functional analysis could help to select phenotype-based therapy in each family member.
FUNDINGThis work was supported by Fondos Europeos de Desarrollo Regional; Ministerio de Economía y Competitividad [SAF2014-58769-P; SAF2017-88116-P]; Comunidad Autónoma de Madrid [B2017/BMD-3738]; Instituto de Salud Carlos III [PI16/00398]; The ERA-Net for Research on Rare Diseases (AC14/00029), Mutua Madrileña and Banco Bilbao Vizcaya Argentaria Foundations, and the Spanish Society of Cardiology Grants.
CONFLICTS OF INTERESTNone declared.
At least 15 genes have been linked so far to LQTS, an inherited primary arrhythmogenic syndrome characterized by a prolonged QT interval that increases the risk of sudden cardiac death due to ventricular fibrillation. LQTS shows marked phenotypic expressivity, which has been mainly attributed to demographic factors and greatly hampers risk stratification in patients and therapeutic decisions. However, in some families, the variable expressivity is due to genetic factors and can be explained by the presence of multiple mutations in the same or different genes (compound or digenic heterozygosity, respectively) or by SNPs.
WHAT DOES THIS STUDY ADD?We functionally analyzed a mutation in the CACNA1C gene encoding p.S1691N Cav1.2 channels found in digenic heterozygosity in a Spanish family. The results demonstrated that this mutation decreased the Ca influx during the plateau phase of the AP, an effect that functionally counterbalances the increase in the Na influx produced by the long QT-associated SCN5A mutation encoding p.R1644H Nav1.5 channels. As a consequence, only the family member carrying the SCN5A mutation exclusively exhibits LQTS type 3. Therefore, phenotypic expression of LQTS could be modulated by genetic factors and the functional analysis of concurrent mutations could help to guide a personalized therapeutic approach.
.
We thank Sandra Sacristán, Lorena Ondo, and Paloma Vaquero for their invaluable technical assistance.