In patients receiving cancer treatment, chemotherapy is potentially cardiotoxic, and ventricular dysfunction secondary to cardiotoxic drugs (VD-CTox) is one of the most common complications.1 Its onset depends on the patient's baseline risk and therefore appropriate stratification is essential. Current recommendations are to consider certain clinical characteristics (age, cardiovascular risk factors, heart disease), other associated cardiotoxic treatments, and, in the case of anthracyclines, the cumulative dose.2 However, some patients develop ventricular dysfunction that is disproportionate to the estimated baseline risk, suggesting an individual susceptibility to the disease. Reports of families with cases of both dilated cardiomyopathy and VD-CTox have been described, indicating a common genetic basis.3 Recently, it has been demonstrated that the truncation variants of the TTN gene are significantly prevalent in patients with VD-CTox and are associated with a higher incidence of adverse events.4 There is little information on other potentially-associated genes, and there are no specific recommendations on genetic study in this context.
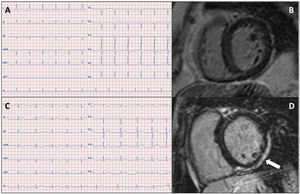
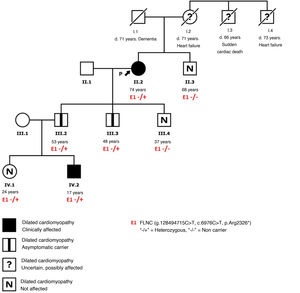
We present the case of a 74-year-old woman with well-controlled hypertension and a history of breast cancer treated with anthracyclines and radiotherapy, with a normal echocardiogram prior to treatment. Ten years after treatment, she developed symptoms of heart failure and was found to have dilated cardiomyopathy with severe left ventricular (LV) systolic dysfunction. Her electrocardiogram showed small R waves in V2-V3, flattened T waves in the inferior leads and V6, and negative T waves in V4-V5 (figure 1A). Coronary angiography was performed, which found healthy coronary arteries, and cardiac magnetic resonance showed no late gadolinium enhancement (figure 1B). Of note in the family history was the presence of cardiovascular disease at a relatively young age on the maternal side, but cardiopathy was not detected on echocardiography in 1 brother and 3 sons (figure 2). To complete the assessment, genetic study was performed using next-generation sequencing, with a dilated cardiomyopathy panel (121 genes), which identified a pathogenic variant in the FLNC gene (p.Arg2326*), previously described in 6 families, in which most of the carriers of the variant had a phenotype of dilated cardiomyopathy and a high incidence of ventricular arrhythmias.
. B: cardiac magnetic resonance of the same patient, without late gadolinium enhancement. C: electrocardiogram of the index patient")
A: electrocardiogram of the index patient (II.2). B: cardiac magnetic resonance of the same patient, without late gadolinium enhancement. C: electrocardiogram of the index patient's nephew (IV.2). D: cardiac magnetic resonance of the same nephew; subepicardial late gadolinium enhancement pattern in the inferior and lateral walls of the left ventricle (arrow).

Family study detected 2 sons who are asymptomatic carriers with preserved ejection fraction and no dilatation, both showing subepicardial late gadolinium enhancement in the lateral wall of the LV, negative T waves in III and aVF on electrocardiography and no arrhythmias on Holter monitoring. One of them has a 17-year-old son, who is a registered football player and a carrier of the variant described. He is asymptomatic but has dilatation and mild dysfunction of the LV, right axis deviation on electrocardiogram (figure 1C) and a subepicardial late gadolinium enhancement pattern on the inferior and lateral walls of the LV (figure 1D), with no arrhythmias on Holter monitoring. His 24-year-old sister is a carrier, but currently has an unaffected phenotype. Both have been recommended to avoid high-intensity sport and undergo close clinical follow-up. The other family members assessed were not carriers of the variant.
The truncation variants of FLNC have been associated with a phenotype of predominantly left-sided dilated/arrhythmogenic cardiomyopathy, characterized by a high incidence of ventricular arrhythmias and a considerable incidence of sudden cardiac death, especially after the age of 40 years, even in the absence of severe systolic dysfunction. Men develop the phenotype earlier than women, and the prognosis, in terms of arrhythmias and sudden cardiac death, is worse than in women.5 For carriers, magnetic resonance is recommended for detection of myocardial fibrosis and Holter monitor for detection of arrhythmias, even if echocardiogram is normal. Competitive sport is not advised in these individuals. Unlike what is described in the literature, in this family, penetrance was incomplete and the phenotype was generally benign. It is possible that the phenotype of some FLNC truncations may be more variable than previously described and that additional environmental triggers may have a strong influence on the severity, such as the chemotherapy and hypertension in the index case or the competitive sport in her nephew, leading to early onset of the disease.
In cancer patients who receive chemotherapy, risk stratification for ventricular dysfunction secondary to treatment remains a challenge. Currently, this is based on clinical markers that cannot predict individual susceptibility. Genetic study can be useful to identify patients with a higher predisposition to VD-CTox, especially when there is a suggestive family history. In addition, it allows early identification of at-risk relatives who could benefit from preventative strategies aimed at delaying disease progression and reducing its complications.
The authors confirm that they obtained informed consent from the patient and their relatives to publish the case, including the images.
FUNDINGNo funding was required.
AUTHORS’ CONTRIBUTIONSM.L. Peña Peña and M.R. Caballero Valderrama were the clinicians treating the patient presented here, performed the literature review, and wrote the manuscript. S. Navarro Herrero and M.P. Serrano Gotarredona reported the cardiac magnetic resonance images and reviewed the manuscript. J.E. López Haldón contributed to the focus of the article and critically reviewed the manuscript.
CONFLICTS OF INTERESTNo conflicts of interest.