
Only about 1 out of every 3 patients with acute myocardial infarction (AMI) achieve low-density lipoprotein cholesterol (LDL-C) values <55mg/dL in the first year. The present study aims to evaluate the impact of early intensive therapy on lipid control after an AMI.
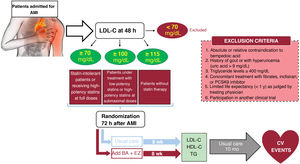
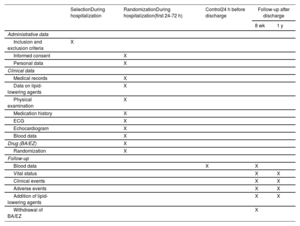
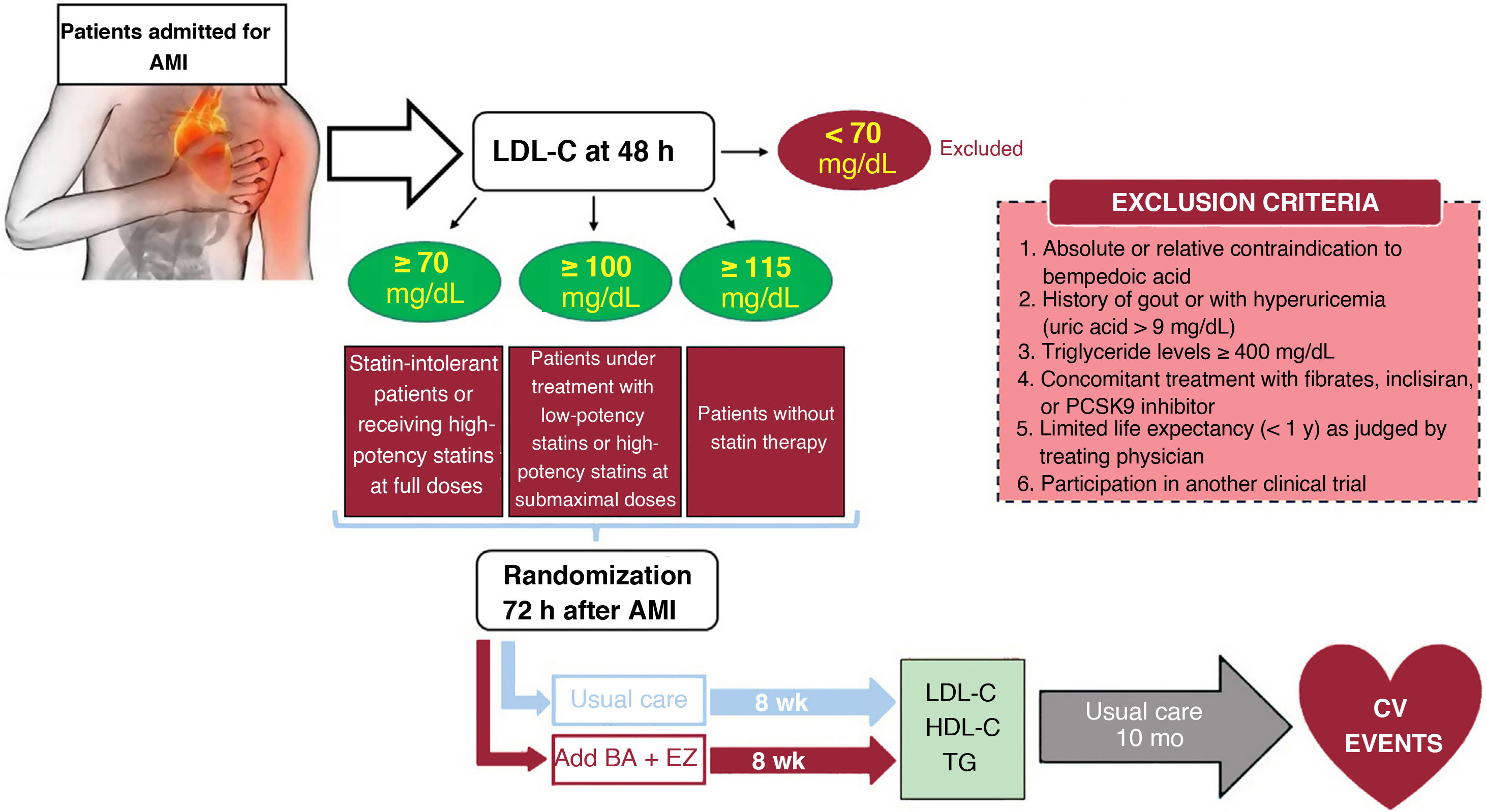
MethodsAn independent, prospective, pragmatic, controlled, randomized, open-label, evaluator-blinded clinical trial (PROBE design) will analyze the efficacy and safety of an oral lipid-lowering triple therapy: high-potency statin+bempedoic acid (BA) 180mg+ezetimibe (EZ) 10mg versus current European-based guidelines (high-potency statin±EZ 10mg), in AMI patients. LDL-C will be determined within the first 48hours. Patients with LDL-C ≥ 115mg/dL (without previous statin therapy), ≥ 100mg/dL (with previous low-potency or high-potency statin therapy at submaximal dose), or ≥ 70mg/dL (with previous high-potency statin therapy at high dose) will be randomly assigned 1:1 between 24 and 72hours post-AMI to the BA/EZ combination or to statin±EZ, without BA. The primary endpoint is the proportion of patients reaching LDL-C <55mg/dL at 8 weeks after treatment.
ResultsThe results of this study will provide novel information for post-AMI LDL-C control by evaluating the usefulness of an early intensive lipid-lowering strategy based on triple oral therapy.
ConclusionsEarly intensive lipid-lowering triple oral therapy vs the treatment recommended by current clinical practice guidelines could facilitate the achievement of optimal LDL-C levels in the first 2 months after AMI (a high-risk period).
Identification numberEudraCT 2021-006550-31.
Keywords
Identify yourself
Not yet a subscriber to the journal?
")
Purchase access to the article
By purchasing the article, the PDF of the same can be downloaded
Price: 19,34 €
Phone for incidents
Monday to Friday from 9am to 6pm (GMT+1) except for the months of July and August, which will be from 9am to 3pm