
Genetic testing is becoming increasingly important for diagnosis and personalized treatments in aortopathies. Here, we aimed to genetically diagnose a group of acute aortic syndrome (AAS) patients consecutively admitted to an intensive care unit and to explore the clinical usefulness of AAS-associated variants during treatment decision-making and family traceability.
MethodsWe applied targeted next-generation sequencing, covering 42 aortic diseases genes in AAS patients with no signs consistent with syndromic conditions. Detected variants were segregated by Sanger sequencing in available family members. Demographic features, risk factors and clinical symptoms were statistically analyzed by Fisher or Fisher-Freeman-Halton Exact tests, to assess their relationship with genetic results.
ResultsAnalysis of next-generation sequencing data in 73 AAS patients led to the detection of 34 heterozygous candidate variants in 14 different genes in 32 patients. Family screening was performed in 31 relatives belonging to 9 families. We found 13 relatives harboring the family variant, of which 10 showed a genotype compatible with the occurrence of AAS. Statistical tests revealed that the factors associated with a positive genetic diagnosis were the absence of hypertension, lower age, family history of AAS and absence of pain.
ConclusionsOur findings broaden the spectrum of the genetic background for AAS. In addition, both index patients and studied relatives benefited from the results obtained, establishing the most appropriate level of surveillance for each group. Finally, this strategy could be reinforced by the use of stastistically significant clinical features as a predictive tool for the hereditary character of AAS.
ClinicalTrials.gov (Identifier: NCT04751058)
Keywords
Acute aortic syndromes (AAS) encompass 3 life-threatening conditions, including acute aortic dissection, intramural hematoma, and penetrating aortic ulcer.1 In Spain, AAS affects 20 to 40 cases/million inhabitants/y and is reported more frequently in men (73%).2 Of these, 70% affect the ascending aorta, 30% the descending portion, and in 78.7%, dissection is the fundamental mechanism.3 Only an approximate 5% of thoracic aortic aneurysms are symptomatic prior to a life-threatening complication that arises unpredictably and presents as sudden cardiac death.4 Therefore, timely diagnosis is essential and saves lives.1 Classically, a number of risk factors have been associated with AAS, including hypertension (76.6%), atherosclerotic disease (27%), use of cocaine (1.8%) or smoking and dyslipidemia.5 Likewise, AAS may be involved in any condition associated with structural changes of the aortic wall, such as connective tissue disorders, previous aneurysms, the presence of bicuspid aortic valve,6 or direct aortic injury following surgery or percutaneous intervention.
AAS may be part of well-characterized genetic syndromes, including Marfan syndrome, Loeys-Dietz syndrome, or vascular Ehlers-Danlos syndrome. In addition, AAS may present as an isolated disease confined to the thoracic aorta (nonsyndromic AAS), and can be sporadic (80% of nonsyndromic AAS4,7) or hereditary, depending on the presence of other affected family members. AAS is a genetically heterogeneous disease with overlapping clinical presentations; hence, patients’ genetic data has become crucial during diagnosis, for the identification of other at-risk relatives8 and personalized patient management, including timing of prophylactic surgical repair.9
Since the introduction of next-generation sequencing (NGS), notable achievements have been made in the discovery of genetic variants and key regulatory molecules associated with AAS.7,10–12 To date, variants in about 40 genes have been associated with AAS, accounting for 20% to 30% of all cases. Autosomal dominant transmission has been proven in the vast majority of patients with the disease, although autosomal recessive and X-linked inheritance have also been observed.13
Aortopathies are more likely to be associated with a genetic cause when the affected individual is younger than 50 years old and/or has a family history of aortic aneurysm or dissection or sudden unexplained death.14 In addition, the presence of some systemic manifestations may be suggestive of syndromic heritable thoracic aortic disease.
In this study, we analyzed the prevalence of genetic variants in a group of nonsyndromic AAS patients admitted to the intensive care unit to explore the clinical usefulness of AAS-associated variants in treatment decision-making and family traceability of this clinically silent and highly morbid disease.
METHODSPatients cohort and surveillanceThis is a prospective observational study of a group of patients consecutively admitted to intensive care unit, University Hospital Virgen del Rocío Seville, with a diagnosis of AAS from October 2017 to January 2020. The study was authorized by the Andalusian Biomedical Research Ethics Platform (PEIBA) (GEN-AOR Internal Code 2436-N-20, Clinical trials [NCT04751058]). Informed consent for the inclusion of the genetic study and for research purposes was collected from all patients or legalguardians when appropriate.
The clinical manifestations considered to be suggestive signs of AAS during initial patient evaluation were the following: signs of sudden, severe chest pain with typical irradiation, hypertension, pulse deficit, difference in blood pressure readings between arms, semiology of aortic insufficiency, signs of ventricular failure, nonspecific electrocardiogram (normal or with left ventricular hypertrophy), normal or with mediastinal widening radiograph and other physical findings that could alert the clinician to the presence of an underlying Marfan syndrome using the Ghent scale. Confirmation of AAS was performed using multislice computed tomography (CT) angiography (angio-CT) (1mm slices), with and without iodinated contrast, for subsequent reconstruction. After confirmation and image analysis, patients were treated according to the recommendations of the European Society of Cardiology Guidelines15 for the affected segment and angiographic conditions.
After clinical evaluation and imaging tests, patients meeting the following criteria were enrolled in the genetic study: a) AAS confirmed by angio-CT; b) aged >16 years and c) absence of signs consistent with syndromic conditions. On the other hand, lifeless entry patients were excluded. Once the patient cohort was selected, the electronic health record was used to collect information on their personal and family history, clinical presentation, and disease course. In this light, the family history was only considered positive if other relatives with aortopathies and/or sudden death were found.
A family segregation analysis was recommended for all cases in which a genetic variant was detected, including likely pathogenic and variants of uncertain significance. After genetic counseling, family members that agreed to undergo clinical examination after a positive or uncertain result in the genetic test were evaluated by echocardiography. Angio-CT was applied during the clinical assessment only after an altered echocardiogram. All positive relatives were referred to the departments of comprehensive care for familial heart diseases and rare diseases of our center.
DNA isolation and quality analysisAutomatic genomic deoxyribonucleic acid (DNA) extraction was performed using a Chemagic 360 instrument (Perkin Elmer, United States). DNA integrity and purity were checked by gel electrophoresis and using both fluorometric and spectrophotometric methods. A detailed description of the complete process can be found in the methods of the supplementary data.
Aortic diseases gene panel designA capture panel (Roche, USA) including 42 genes (table 1 of the supplementary data) related to different aortic diseases was used. The selection of captured genes was based on current guidelines, public databases, and expert consensus. Further details are shown in the methods of the supplementary data.
Library preparation and sequence data generationOne microgram of genomic DNA was used for each library preparation using the SeqCap EZ Library SR version 5.1 (Roche, USA) according to the manufacturer's protocol with minor modifications (methods of the supplementary data). Sequencing was performed in the Illumina NextSeq500 platform (Illumina, USA).
Bioinformatic analysisData analysis was performed using a previously validated pipeline16 with some modifications (methods of the supplementary data).
Variant prioritization and pathogenicity assessmentVariants were prioritized based on established clinical and genetic criteria (methods of the supplementary data). Finally, only variants classified as pathogenic, likely pathogenic or variants of unknown significance were reported.
Statistical analysisContinuous variables are expressed as mean±standard deviation, while categorical variables are expressed as absolute numbers and percentages. All cases were classified in 2 groups: a) individuals with clearly pathogenic or likely pathogenic variants, and b) individuals with no variants or with clinically uncertain variants. Differences between both groups for categorical variables were estimated by Fisheŕs exact test, except for contingency tables larger than 2x2, for which the Fisher-Freeman-Halton Exact test was used. The obtained results were considered statistically significant for 2-sided P-values <.05. All statistical analyses were performed using the IBM SPSS Statistics for Windows, Version 25.0 (IBM Corp., USA).
To evaluate the diagnostic accuracy of significant clinical variables and their combination to be used as heritability-prediction tools, the following attributes were calculated17,18: true positive rate (or sensitivity), true negative rate (or specificity), false positive rate, false negative rate, and the odds ratio (OR), representing the predicted probability that a case with a given clinical variable is genetically positive against the probability that a case without that clinical variable has a positive genetic test. An OR greater than 1 implies a fold-increased odd that a positive genetic case manifests a given clinical variable vs it does not. Of note, cases classified as uncertain were not included in these calculations.
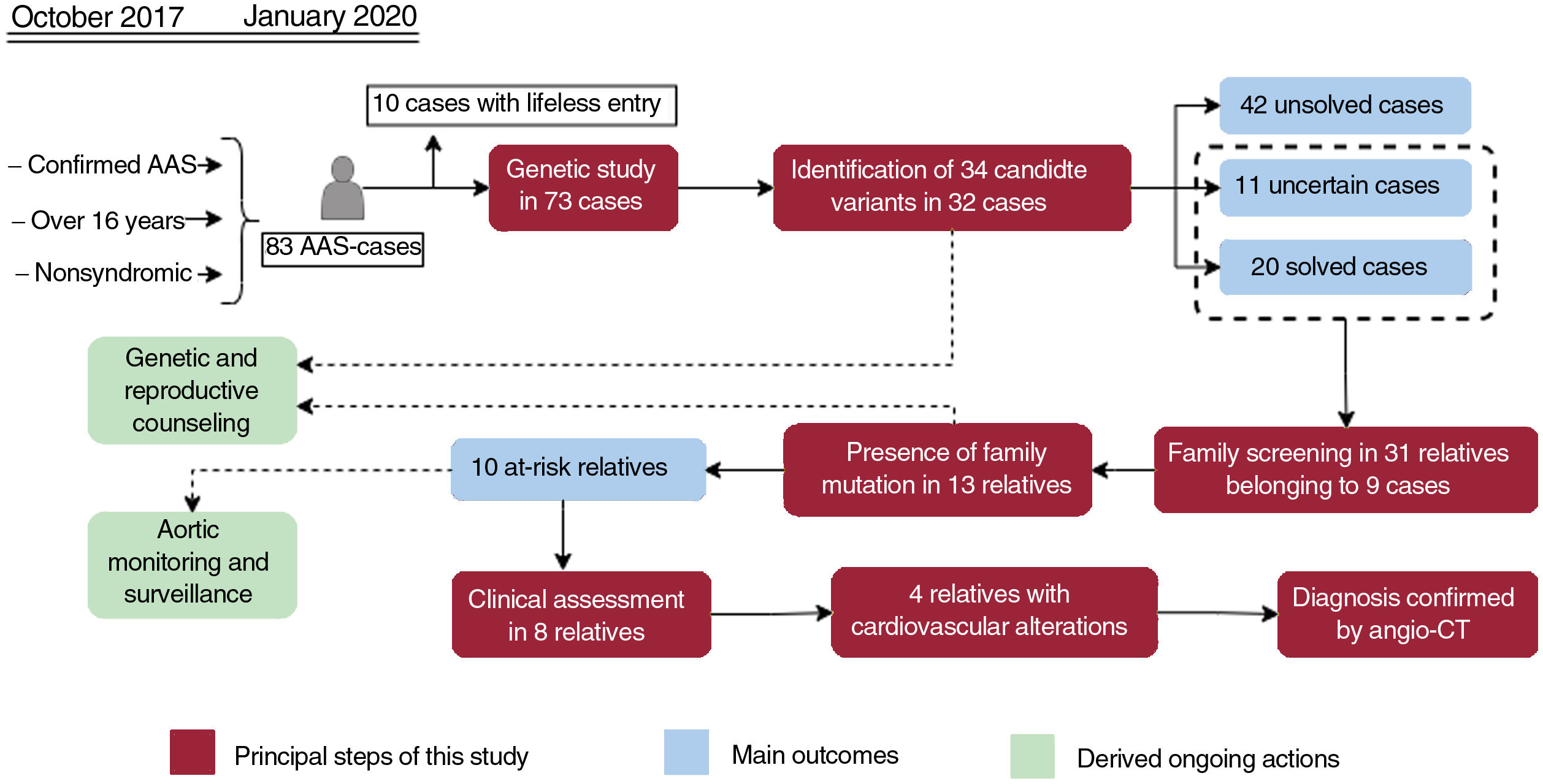
RESULTSDemographic and clinical characteristics of acute aortic syndrome casesFrom October 2017 to January 2020, 83 AAS patients admitted to the intensive care unit met the inclusion criteria for the genetic study. However, 10 of them died in the first hours without the possibility of obtaining DNA samples, leaving a total of 73 patients for the study. Table 1 shows the general clinical characteristics of this group of patients, while detailed information for each of the cases is shown in table 2 of the supplementary data. The median age was 57 years with an approximate male/female ratio of 3:1. Based on Standford classification, 63% of patients were Stanford A and 37%, Stanford B cases. Dissection (74%) was the most frequent clinical form. Hypertension and smoking were the most common risk factors, being even more represented in patients with aortic dissection type B (70.3% and 81.4%, respectively). In 57 patients, the first symptom was pain, accompanied by a hypertensive crisis in 27 patients. Sixty patients (82.2%) underwent surgical treatment, 46.2% on an emergency and/or urgent basis. Of note, 47.9% of the patients had an aortic diameter less than 5cm.
Characteristics of the study group
| Number/mean±SD | Percentage/intervals | P | |
|---|---|---|---|
| Patients description | |||
| Patients in total | 73 | 100 | - |
| Age at the AAS presentationa | 57±13.16 | 16-81 | - |
| <55 y | 33 | 45.2 | < .001c |
| Male | 54 | 74 | .241 |
| Female | 19 | 26 | |
| Geography, race | |||
| Andalusia | 70 | 95.8 | .557 |
| Other | 3 | 4.2 | |
| Caucasian race | 73 | 100 | - |
| Type of clinical presentation | |||
| Acute (<15 d) | 54 | 74 | 1.000b |
| Subacute (> 15 d) | 7 | 9.6 | |
| Chronic (> 1 mo) | 12 | 16.4 | |
| Stanford A | 46 | 63 | .278 |
| Stanford B | 27 | 37 | |
| Diameter of the aorta, cma | 4.57±1.5 | 2.5-12.5 | - |
| Type | |||
| Dissection | 52 | 74 | .562b |
| Penetrating ulcer | 5 | 6.8 | |
| Intramural hematoma | 7 | 9.6 | |
| Rupture aneurysm | 2 | 2.7 | |
| Aneurysm | 7 | 9.6 | |
| Risk factors | |||
| Smoking | 44 | 60.3 | .424 |
| Hypertension | 42 | 57.3 | .007c |
| Cardiac surgery | 3 | 4.1 | .557 |
| DM | 5 | 6.8 | .610 |
| COPD | 8 | 11 | .432 |
| Family history | 15 | 20.5 | .003c |
| Hyperlipidemia | 19 | 26 | .241 |
| Drug abuse | 15 | 20.5 | .267 |
| Bicuspic valve | 2 | 2.7 | 1.000 |
| Clinical symptoms | |||
| Pain | 57 | 78.1 | .009c |
| Hypertensive crisis | 28 | 38.4 | 1.000 |
| Pulse deficit | 23 | 31.5 | 1.000 |
| Shock | 13 | 17.8 | .326 |
| Acute renal failure | 21 | 28.8 | .393 |
| Cardiac tamponade | 6 | 8.2 | .663 |
| Visceral ischemia | 12 | 16.4 | 1.000 |
| Syncope | 14 | 19.4 | .744 |
| Treatment | |||
| Medical treatment | 13 | 17.8 | .742 |
| Surgical treatment | 60 | 82.2 | |
| Open surgery | 40 | 66.6 | .371 |
| Endovascular | 20 | 27.4 | |
| Treatment criteria | |||
| Emergent | 28 | 38.4 | .833b |
| Urgent | 13 | 17.8 | |
| Delayed | 19 | 26 | |
| ICU staya | 11±11.2 | 1-51 | - |
| Hospital staya | 21±17.5 | 1-70 | - |
| AAS-related deaths | 14 | 19.2 | .747 |
AAS, acute aortic syndrome; DM, diabetes mellitus; COPD, chronic obstructive pulmonary disease; ICU, intensive care unit; SD, standard deviation.
The data are expressed as the mean±standard deviation and intervals for continuous variables(a) and absolute numbers and percentages for categorical variables. P-values show the significance obtained by applying the statistical test (Fisher exact test or Fisher-Freeman-Halton Exact Test (b), as appropriate) to relate the genetic results with categorical variables, with those marked with a letter c being statistically significant (c).
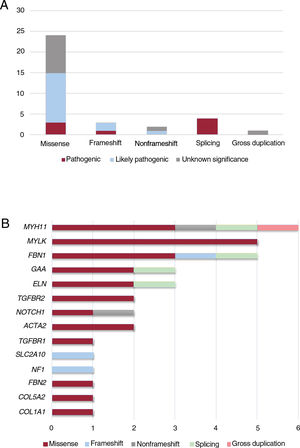
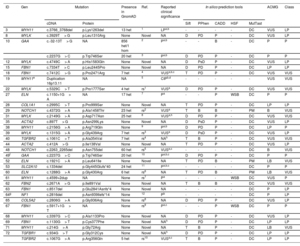
Our NGS application allowed us to cover 97.1% of target bases, resulting in a mean coverage of 357.9x and a percentage of reads mapped on target of 83.4%. All coding regions corresponding to genes associated with aortic diseases (table 1 of the supplementary data), were covered> 20x. NGS data analysis led to the detection of 34 heterozygous candidate genetic variants in a total of 32 patients (table 2 and figure 1), including 24 missense, 4 splicing, 3 frameshift, 2 nonframeshift deletions, and 1 copy number variation (figure 2A).
Likely causative mutations and variants of unknown significance identified in the study cohort
| ID | Gen | Mutation | Presence in GnomAD | Ref. | Reported clinical significance | In silico prediction tools | ACMG | Class | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cDNA | Protein | Sift | PPhen | CADD | HSF | MutTast | |||||||
| 3 | MYH11 | c.3766_3768del | p.Lys1263del | 13 het | 1 | LPa,b | - | - | - | - | DC | VUS | LP |
| 8 | MYLK | c.3929T> G | p.Leu1310Arg | None | Novel | NA | D | PD | P | - | DC | VUS | LP |
| 10 | GAA | c.-32-13T> G | NA | 856 het/1 hom | 2 | Pa,b | - | - | B | - | DC | P | P |
| c.2237G> C | p.Trp746Ser | 20 het | 3 | Pa,b | D | PD | P | - | DC | P | P | ||
| 12 | MYLK | c.4749C> A | p.His1583Gln | None | Novel | NA | D | PsD | P | - | DC | VUS | LP |
| 15 | FBN1 | c.7334T> C | p.Leu2445Pro | None | Novel | NA | D | PD | P | - | DC | LP | LP |
| 18 | FBN1 | c.7412C> G | p.Pro2471Arg | 7 het | 4 | VUSa,b,c | T | PD | P | - | DC | VUS | VUS |
| 19 | MYH11e | Duplication 16p13.11 | NA | NA | 5 | CIPb,d | - | - | - | - | - | VUS | VUS |
| 22 | MYLK | c.5329C> T | p.Pro1777Ser | 4 het | rs1 | VUSa | D | PD | P | - | DC | VUS | VUS |
| 27 | ELN | c.1150+1G> A | NA | 17 het | 7 | Pa | - | - | P | WSB | DC | P | P |
| 28 | COL1A1 | c.2995C> T | p.Pro999Ser | None | Novel | NA | T | PD | P | - | DC | LP | LP |
| 29 | NOTCH1 | c.4372G> A | p.Ala1458Thr | 23 het | rs2 | VUSa | T | B | B | - | PM | B | VUS |
| 31 | MYLK | c.2149G> A | p.Asp717Asn | 25 het | 8 | VUSa,h | D | PD | P | - | DC | VUS | VUS |
| 35 | ACTA2 | c.897T> G | p.Asn299Lys | None | Novel | NA | D | PsD | P | - | DC | VUS | LP |
| 38 | MYH11 | c.2156G> A | p.Arg719Gln | None | 9 | Pa,b | D | PD | P | - | DC | LP | P |
| 39 | MYLK | c.1315G> A | p.Gly439Arg | 7 het | rs3 | VUSa | D | PsD | P | - | DC | VUS | LP |
| 40 | TGFBR2 | c.1061C> T | p.Ala354Val | 7 het | rs4 | NAh | T | B | P | - | DC | VUS | VUS |
| 44 | ACTA2 | c.412A> G | p.Ile138Val | None | Novel | NA | T | PD | P | - | DC | VUS | LP |
| 48 | NOTCH1 | c.2263_2265del | p.Asn755del | 60 het | rs5 | VUSa,c | - | - | - | - | DC | B | VUS |
| 49f | GAA | c.2237G> C | p.Trp746Ser | 20 het | 10 | Pa,b,c | D | PD | P | - | DC | P | P |
| 52 | ELN | c.1921C> A | p.Leu641Ile | None | Novel | NA | T | PD | B | - | PM | LB | VUS |
| 53 | SLC2A10 | c.1334del | p.Gly445Glufs*40 | 24 het | 11 | Pa,b,c | - | - | - | - | DC | P | P |
| 60 | ELN | c.1288G> A | p.Gly430Arg | 6 het | rs6 | NA | T | PD | B | - | PM | LB | VUS |
| 61 | MYH11 | c.4599+2dup | NA | None | rs7 | Pa | - | - | - | WSB | DC | VUS | P |
| 62 | FBN2 | c.2671A> G | p.Ile891Val | None | Novel | NA | T | B | B | - | DC | VUS | VUS |
| 63 | FBN1 | c.8517del | p.Glu2841Asnfs*4 | None | Novel | NA | - | - | - | - | DC | P | LP |
| 64g | NF1 | c.2816del | p.Asn939Ilefs*14 | None | 12 | Pb | - | - | - | - | DC | P | LP |
| 65 | COL5A2 | c.2806G> A | p.Gly936Arg | None | rs8 | NA | D | PD | P | - | DC | VUS | LP |
| 67 | FBN1 | c.5917+1G> T | NA | None | rs9 | Pa,c | - | - | P | WSB | DC | P | P |
| 68 | MYH11 | c.3397G> C | p.Ala1133Pro | None | Novel | NA | D | PD | P | - | DC | VUS | LP |
| 69 | FBN1 | c.1130G> T | p.Cys377Phe | None | Novel | NAh | D | PD | P | - | DC | LP | LP |
| 71 | MYH11 | c.214G> A | p.Gly72Arg | None | Novel | NA | T | B | P | - | DC | LB | VUS |
| 72 | TGFBR1 | c.934G> T | p.Gly312Cys | None | Novel | NAh | D | PD | P | - | DC | LP | LP |
| TGFBR2 | c.1067G> A | p.Arg356Gln | 5 het | rs10 | VUSa,c | T | B | P | - | DC | LP | LP | |
ACMG, variant classification based on American College of Medical Genetics and Genomics criteria; B, benign; CADD, combined annotation dependent depletion (Variants were considered pathogenic with a score ≥ 15); CIP, conflicting interpretation of pathogenicity; D, damaging; DC, disease-causing. Het, heterozygotes; Hom, homozygotes; HSF, human splicing finder; ID, identification number; LB, likely benign; LP, likely pathogenic; MutTast, mutation taster; NA, not available; Our Class, variant classification based on all the collected information; P, pathogenic; PD, probably damaging; PM, polymorphism; Pphen, Polyphen-2; PsD, possibly damaging; Ref, references; rs1, rs748200926; rs2, rs200495793; rs3, rs190877071; rs4, rs376752333; rs5, rs587778559; rs6, rs274470120; rs7, rs794728677; rs8, rs1471655044; rs9, rs363808; rs10, rs727504292; T, tolerated; VUS, variant of unknown significance; WSB, wild-type splicing site broken.
All detected variants were found in heterozygous state. The reported clinical significance was checked both in the databases ClinVar (a), LOVD (b) and HGMD (c), and the literature (d).

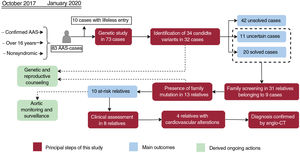
Central illustration. Flowchart of patients, family screening, and genetic testing outcomes. Orange boxes reflect the principal steps performed in our workflow, while the main outcomes are shown in red boxes. The ongoing actions derived as a consequence of this study are indicated in green boxes. AAS, acute aortic syndrome; Angio-CT, multislice computed tomography angiography.

Classification of the 34 identified variants associated with aortic disease in our cohort. A: type of variants according to consequence type. B: distribution of variant types, including missense, frameshift, nonframeshift, splicing and gross duplication in genes associated with aortic disease in our cohort.
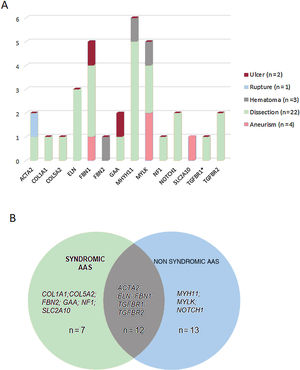
These variants were found in 14 different genes, with MYH11 being the most prevalent mutated gene followed by MYLK and FBN1 (figure 2B). Although these genes are involved in a wide spectrum of molecular pathways (table 1 of the supplementary data), most of them (22 cases, 69%) were associated with aortic dissection in our cohort (figure 3A). In all cases, our results were compatible with an autosomal dominant inheritance pattern except for family 10, for which compound heterozygosis of 2 changes in GAA was considered the most likely disease cause.

Genotype-phenotype correlations. A: representation of the type of aortic disease associated with the genes identified in this study. B: number of cases harboring variants in genes associated with syndromic, nonsyndromic or to both forms of aortic disease. The asterisk denotes a variant in the TGFBR1 gene present in 1 of the cases harboring a concomitant variant in TGFBR2.
Approximately, 40.7% of patients (n=13) harbored genetic variants in genes previously associated with familial, nonsyndromic forms, 21.8% (n=7) with syndromic conditions, and 37.5% (n=12) carried variants in genes associated with both syndromic and nonsyndromic forms (figure 3B). Interestingly, 14 a priori nonsyndromic cases harbored pathogenic or likely pathogenic variants in genes previously associated with a potential syndromic aortic disease. Consequently, patients 10, 53, 64, and 69 were diagnosed with Pompe disease, arterial tortuosity syndrome, neurofibromatosis, and Marfan syndrome, respectively, and patients 28 and 65 were diagnosed with Ehler-Danlos syndrome. This allowed us to reclassify 18.7% of cases to a syndromic aortopathy, which has important implications for patient and family management.
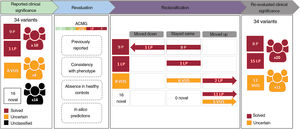
Assessment of the clinical significance of identified variantsTen of the 34 identified variants had previously been reported as pathogenic or likely pathogenic in public databases and/or the literature, whereas 8 were previously described as variants of unknown significance, and 16 were novel without any clinical association so far (table 2 and figure 4). A re-evaluation of variants was performed according to the previous clinical associations, the allele count in gnomAD, the in silico pathogenicity predictions and the American College of Medical Genetics and Genomics criteria, resulting in the reclassification of 19 variants (figure 4). This resulted in the identification of 11 variants of unknown significance in 11 cases (15.07%) and 23 pathogenic or likely pathogenic variants in 21 patients, although only 20 cases were considered solved (27.39%), since a unique heterozygous variant in the autosomal recessive gene GAA was found in patient 49. If a clinical association is confirmed for these variants, the diagnostic rate of this study could reach up to 42.46%.

Workflow of variant re-evaluation, classification, and clinical interpretation. ACMG, variant classification based on American College of Medical Genetics and Genomics criteria; B, benign; LB, likely benign; LP, likely pathogenic; P, pathogenic; VUS, variant of unknown significance.
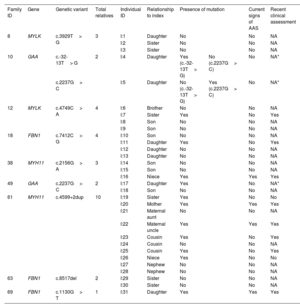
Family members belonging to 9 families with detected candidate variants were willing to be studied (table 3 and figure 1). We confirmed the presence of the detected variants in 13 out of 31 relatives analyzed by Sanger sequencing. Three (I:4, I:5 and I:17) of the 13 relatives were carriers of genetic variants associated with an autosomal recessive trait and hence their genotypes were not compatible with a AAS phenotype. The remaining 10 individuals harbored genetic variants compatible with an autosomal dominant inheritance and therefore their genotypes were consistent with the occurrence of AAS.
Family segregation of the genetic variants identified in the 9 patients with available relatives
| Family ID | Gene | Genetic variant | Total relatives | Individual ID | Relationship to index | Presence of mutation | Current signs of AAS | Recent clinical assessment | |
|---|---|---|---|---|---|---|---|---|---|
| 8 | MYLK | c.3929T> G | 3 | I:1 | Daughter | No | No | NA | |
| I:2 | Sister | No | No | NA | |||||
| I:3 | Sister | No | No | NA | |||||
| 10 | GAA | c.-32-13T> G | 2 | I:4 | Daughter | Yes (c.-32-13T> G) | No (c.2237G> C) | No | NA* |
| c.2237G> C | I:5 | Daughter | No (c.-32-13T> G) | Yes (c.2237G> C) | No | NA* | |||
| 12 | MYLK | c.4749C> A | 4 | I:6 | Brother | No | No | NA | |
| I:7 | Sister | Yes | No | Yes | |||||
| I:8 | Son | No | No | NA | |||||
| I:9 | Son | No | No | NA | |||||
| 18 | FBN1 | c.7412C> G | 4 | I:10 | Son | No | No | NA | |
| I:11 | Daughter | Yes | No | Yes | |||||
| I:12 | Daughter | No | No | NA | |||||
| I:13 | Daughter | No | No | NA | |||||
| 38 | MYH11 | c.2156G> A | 3 | I:14 | Son | No | No | NA | |
| I:15 | Son | No | No | NA | |||||
| I:16 | Niece | Yes | Yes | Yes | |||||
| 49 | GAA | c.2237G> C | 2 | I:17 | Daughter | Yes | No | NA* | |
| I:18 | Son | No | No | NA | |||||
| 61 | MYH11 | c.4599+2dup | 10 | I:19 | Sister | Yes | No | No | |
| I:20 | Mother | Yes | Yes | Yes | |||||
| I:21 | Maternal aunt | No | No | NA | |||||
| I:22 | Maternal uncle | Yes | Yes | Yes | |||||
| I:23 | Cousin | Yes | No | Yes | |||||
| I:24 | Cousin | No | No | NA | |||||
| I:25 | Cousin | Yes | No | Yes | |||||
| I:26 | Niece | Yes | No | No | |||||
| I:27 | Nephew | No | No | NA | |||||
| I:28 | Nephew | No | No | NA | |||||
| 63 | FBN1 | c.8517del | 2 | I:29 | Sister | No | No | NA | |
| I:30 | Sister | No | No | NA | |||||
| 69 | FBN1 | c.1130G> T | 1 | I:31 | Daughter | Yes | Yes | Yes | |
AAS, acute aortic syndrome; ID, identification number; NA, not applicable.
All individuals harboring the mutations are heterozygous. Individuals with current signs of AAS showed cardiovascular alterations according to the latest imaging tests. A recent clinical assessment was offered to patients in whom a causative genotype was detected, which was not applicable for wild-type patients and carriers of autosomal recessive variants (*).
A clinical reassessment was offered to the 10 relatives with a positive result, but only 8 have attended to date. Among them, 4 patients already showed cardiovascular alterations: a) individual I:16 (16 years) showed a resting cardiac electrical activity with QTc in upper limits (464ms); b) individual I:20 (67 years) showed mild dilatation of the root (37mm) and ascending aorta (40mm), and a grade II aortic insufficiency; c) individual I:21 (63 years) showed mild dilatation of the root (43mm) and ascending aorta (38mm), and a mild diffuse calcified atheromatosis; and d) individual I:31 (15 years) showed a mild dilatation of pulmonary artery. Remarkably, with the exception of individual I:7, all relatives with positive genetic results and no current signs of aortopathies were younger than the index patient of their family. Indeed, 11 out of 13 relatives with a finding of familial variants are currently of reproductive age (< 50 years).
Correlation of genetic results with clinical variablesStatistical tests showed significant differences between patients with a clear genetic diagnosis and those with no causative or uncertain variants, for the following clinical variables: hypertension, patient age, family history, and pain (table 1 and figure 5). The absence of hypertension, an age <55 years old, having a family history of AAS and without pain as a first symptom, significantly correlated with the detection of pathogenic or likely pathogenic genetic variants and, therefore, with hereditary cases.
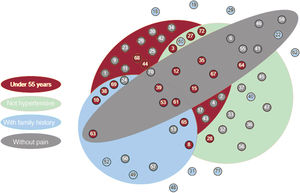
, variants of unknown significance (blue), or no candidate variants (grey), were found.")
Association between hereditary acute aortic syndrome cases and significant clinical features. Each circle corresponds to 1 of the families under study, labeled with its identification number. The color of the circles indicates whether pathogenic or likely pathogenic variants (red), variants of unknown significance (blue), or no candidate variants (grey), were found.
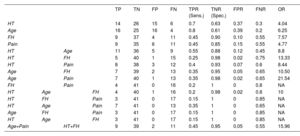
All solved cases were included in at least 1 of these 4 statistically significant groups whereas the combination of the 4 significant variables encompassed only 3 of these cases (figure 5). Indeed, table 4 shows that, while sensitivity was highest for age and hypertension (a true positive rate of 70% and 80%, respectively), family history and pain were the most specific (a true negative rate of 90% and 85%, respectively). The combination of these clinical variables allowed us to obtain a balance between sensitivity and specificity, as well as to improve their predictive ability according to the calculation of OR. If we analyze the highest values of this attribute, the combination of age and pain (OR=21.54) and the combination of hypertension and family history (OR=13.33) would be the sets with a greatest predictive strength, although their sensitivities were decreased (35% and 25%, respectively). However, the joint use of these 2 combinations allowed us to maintain a high predictive value (OR=15.96) without excessively reducing sensitivity (45%) and ensuring specificity (95%).
Attributes calculated to measure the diagnostic accuracy of statistically significant clinical variables
| TP | TN | FP | FN | TPR (Sens.) | TNR (Spec.) | FPR | FNR | OR | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HT | 14 | 26 | 15 | 6 | 0.7 | 0.63 | 0.37 | 0.3 | 4.04 | |||
| Age | 16 | 25 | 16 | 4 | 0.8 | 0.61 | 0.39 | 0.2 | 6.25 | |||
| FH | 9 | 37 | 4 | 11 | 0.45 | 0.90 | 0.10 | 0.55 | 7.57 | |||
| Pain | 9 | 35 | 6 | 11 | 0.45 | 0.85 | 0.15 | 0.55 | 4.77 | |||
| HT | Age | 11 | 36 | 5 | 9 | 0.55 | 0.88 | 0.12 | 0.45 | 8.8 | ||
| HT | FH | 5 | 40 | 1 | 15 | 0.25 | 0.98 | 0.02 | 0.75 | 13.33 | ||
| HT | Pain | 8 | 38 | 3 | 12 | 0.4 | 0.93 | 0.07 | 0.6 | 8.44 | ||
| Age | FH | 7 | 39 | 2 | 13 | 0.35 | 0.95 | 0.05 | 0.65 | 10.50 | ||
| Age | Pain | 7 | 40 | 1 | 13 | 0.35 | 0.98 | 0.02 | 0.65 | 21.54 | ||
| FH | Pain | 4 | 41 | 0 | 16 | 0.2 | 1 | 0 | 0.8 | NA | ||
| HT | Age | FH | 4 | 40 | 1 | 16 | 0.2 | 0.98 | 0.02 | 0.8 | 10 | |
| HT | FH | Pain | 3 | 41 | 0 | 17 | 0.15 | 1 | 0 | 0.85 | NA | |
| HT | Age | Pain | 7 | 41 | 0 | 13 | 0.35 | 1 | 0 | 0.65 | NA | |
| Age | FH | Pain | 3 | 41 | 0 | 17 | 0.15 | 1 | 0 | 0.85 | NA | |
| HT | Age | FH | 3 | 41 | 0 | 17 | 0.15 | 1 | 0 | 0.85 | NA | |
| Age+Pain | HT+FH | 9 | 39 | 2 | 11 | 0.45 | 0.95 | 0.05 | 0.55 | 15.96 |
FH, family history; FN, false negatives; FNR, false negative rate; FP, false positives; FPR, false positive rate; HT, hypertension; OR, odds ratio; TN, true negatives; TNR, true negative rate or specificity (Spec.); TP, true positives; TPR, true positive rate or sensitivity (Sens.).
The effective sample size was 61 since cases with variants of unknown significance were not included in calculations.
Aortic disease is a major diagnostic challenge due to its silent nature; however, its complications constitute an important source of extreme severity and mortality. Because more than 20% of patients report a positive family history, AAS genetics has been extensively investigated as a potential avenue for both diagnosis and risk stratification.
In this study, we used NGS-based multigene testing as a screening method to identify disease-related genetic variants in the context of the molecular diagnosis of aortic diseases. The identification of marked differences in the diagnostic yield (3% to 37%) is common in this group of diseases,10,11,19 which has been attributed to differences in patient inclusion criteria, variant classification rules, and the selection of genes to be included in the study. In line with previously reported data,10,11,19 we achieved a diagnostic rate of 27.39%, which may increase to 42.46% if variants classified as variant of unknown significance are eventually upgraded to likely pathogenic.
Bioinformatic analysis enabled us to detect 34 heterozygous candidate genetic variants in 14 AAS-associated genes, with MYH11 being the most prevalent mutated gene followed by MYLK and FBN1, which is consistent with previously reported prevalence data.19,20 In contrast, genetic variants in the ACTA2 gene are underrepresented (6.2%) in our population in comparison with previous reports (12%-21%),21 which could indicate a less relevant role of this gene in our population, or a bias due to diverse inclusion criteria.
Establishing genotype-phenotype correlations is especially hampered in these disorders due to the wide spectrum of physical manifestation, suggesting the involvement of multifactorial genetic or environmental risk factors.10,22 Our results reinforce the notion that a hypothesis-free approach regardless of clinical assumptions involving genes carefully selected by experts considerably improves the genetic management of these patient.23 Moreover, most life-threatening autosomal dominant diseases notoriously hamper diagnosis prior to complications.4 Thus, a common limitation is sample size and the availability of affected family members to undergo conclusive segregation studies. Indeed, in this study, familial segregation was only performed in 9 out of 13 families, highlighting the importance of conducting periodic reassessments to refine the categorization of variants. This arduous task is especially relevant when concomitant genetic defects are detected.24 Of note, 2 likely pathogenic variants in TGFBR1 and TGFBR2, respectively, were found in patient 72; however, additional studies are needed to discern the degree of involvement of each of them in the appearance or severity of the disease.
In addition, beyond clarifying genotype-phenotype correlations and achieving a genetic diagnosis of index patients, genetic testing in AAS has a crucial role in determining familial risk.25 Asymptomatic older and younger at-risk relatives of an affected individual should be promptly evaluated to clarify their genetic status and to establish the appropriate level of surveillance. Genetic screening for familial aortopathy in 31 family members identified a genotype compatible with the development of AAS in 10 individuals who are currently being monitored and treated upon symptom appearance (I:16, I:20, I:21 and I:31). Furthermore, 3 additional individuals (I:4, I:5 and I:17) carried potential AAS-causing variants in the context of an autosomal recessive disorder. Therefore, a total of 13 individuals may also benefit from reproductive genetic counseling and preimplantation genetic tests, which is especially relevant considering that most of the studied family members are of reproductive age. Furthermore, the identification of at-risk relatives prevented the monitoring of 15 individuals lacking the disease-causing variant, reducing associated costs, and providing a sense of relief for these patients. Nevertheless, the absence of a genetic variant with unknown clinical significance in an individual (ie family 18) is not enough to be exempt that individual from surveillance.20
Importantly, the establishment of genotype-phenotype correlations not only allows us to anticipate the symptoms in patients harboring causative genetic variants, but also to predict the hereditary character of AAS even when the underlying genetic defect has not yet been identified. In this study, 4 clinical characteristics were significantly associated with the presence of pathogenic variants. Whereas clinical variables including normal blood pressure, early presentation of the disease and a positive family history have been previously associated with hereditary AAS,24,26 this study also points to the absence of pain as an indicator of a heritable thoracic aortic disease. Indeed, we suggest that the combination of the clinical variables age and pain, together with the combination of hypertension and family history, may be applied as a useful predictive tool for heritability. Remarkably, other clinical variables commonly associated with genetic results, such as the type of aortic dissection,27 were not associated in this study, suggesting differences among populations or a bias related to our limited sample size.
CONCLUSIONSIn summary, the implementation of this NGS strategy guided the clinical management of our patients with aortic diseases by preventing emergency events for them and their relatives and providing appropriate genetic and reproductive counseling. As genetic testing becomes more widespread and accurate, a comprehensive library of pathogenic genetic variants could be developed, improving risk stratification, the possibility of genetically informed personalized surgical interventions, discrimination between AAS from other serious conditions, and management of clinically challenging AAS cases.
FUNDINGThis work was supported by Instituto de Salud Carlos III (ISCIII), Spanish Ministry of Economy and Competitiveness, Spain and co-funded by the European Union (ERDF, “A way to make Europe”) [PI18-00612; PI19/01550; PI21-00244; IMP/00009], Regional Ministry of Health and Families of the Autonomous Government of Andalusia [PEER-0501-2019; PEER-0470-2019], Regional Ministry of Economic Transformation, Industry, Knowledge and Universities of Andalusia [P20_00887] and the Foundation Isabel Gemio/Foundation Cajasol [FGEMIO-2019-01]. NBG is supported by a fellowship RH-0118-2020, which has been funded by the Regional Ministry of Health and Families of the Autonomous Government of Andalusia.
AUTHORS’ CONTRIBUTIONSA.M. Puppo Moreno, and S. Borrego conceived the study. A.M. Puppo Moreno, S. Borrego, N. Bravo-Gil, and C. Méndez-Vidal designed the study and drafted the manuscript. A.M. Puppo Moreno, A. Adsuar Gómez, F. Tadeo Gómez Ruiz, C. Jiménez de Juan, R. Martín Bermúdez, J.M. López Sánchez, S. Martín Sastre, and M. Fernández Caro collected clinical data and performed clinical management. A.M. Puppo Moreno, P. Gallego and S. Borrego interpreted clinical data. N. Bravo-Gil, C. Méndez-Vidal and R.M. Fernández García analyzed and interpreted genomic data. S. Borrego, N. Bravo-Gil, and C. Méndez-Vidal collected genomic data. A. Adsuar Gómez, F. Tadeo Gómez Ruiz, C. Jiménez de Juan, R.M. Fernández García, R. Martín Bermúdez, J.M. López Sánchez, S. Martín Sastre, M. Fernández Caro, and P. Gallego revised the manuscript critically for important intellectual content. All authors approved the final version to be published. A.M. Puppo Moreno, N. Bravo-Gil and C. Méndez-Vidal should be considered as joint first authors.
CONFLICTS OF INTERESTAll authors report no disclosures.
- •
Genetic testing has become a crucial element during the diagnosis, management, surveillance, risk stratification and familial evaluation of AAS patients.
- •
To date, variants in about 40 genes have been associated with AAS.
- •
Specific genotype-phenotype correlations continue to emerge, allowing for more precise and individualized management and treatment of patients with AAS.
- •
The identification of novel variants associated with AAS, expanding our knowledge of the genetic background of this disorder.
- •
The description of how the genetic findings can contribute to strategies to prevent and treat AAS.
- •
The possibility to predict the hereditable character of AAS through the assessment and combination of clinical features such as hypertension, age, pain, and family history.
Supplementary data associated with this article can be found in the online version, at https://doi.org/10.1016/j.rec.2022.10.005