Congenital long QT syndrome is mainly caused by mutations in the KCNQ1, KCNH2 and SCN5A genes. The aim of this study was to investigate the prevalence of mutations in these three genes in patients with long QT syndrome or idiopathic ventricular fibrillation seen at our center. The study included nine patients with long QT syndrome and four with idiopathic ventricular fibrillation. The first-degree relatives of genotype-positive probands were also investigated. Missense mutations were found in seven patients with long QT syndrome and two with idiopathic ventricular fibrillation. Overall, 71.4% of mutations were in KCNH2 and 28.6% were in SCN5A. No mutations were found in KCNQ1. Only two mutations had been previously observed. Mutations were also found in six of the 19 relatives studied. In conclusion, our initial experience shows that genetic testing had a high sensitivity for diagnosing long QT syndrome. Mutations were found most frequently in the KCNH2 gene.
Keywords
Long QT syndrome (LQTS) is a cardiac channelopathy that can lead to sudden death caused by ventricular arrhythmias. Hundreds of mutations associated with this condition have been described in 12 different genes, mainly encoding sodium and potassium channels.1,2 Approximately 75% of the mutations described in LQTS are located in 3 genes: KCNQ1 (potassium channel), KCNH2 (potassium channel), and SCN5A (sodium channel).1 In 25%–30% of patients with LQTS, complete sequencing of all the genes with known mutations fails to yield a genetic diagnosis.3
Since LQTS has a penetrance of between 25% and 90%, patients with the disease may nevertheless have a normal electrocardiogram (ECG). In the event of cardiac arrest, these patients may be classified as having idiopathic ventricular fibrillation (IVF). Although the etiology of LQTS is not restricted to channelopathies, the potential usefulness of testing for mutations in the genes implicated in LQTS has recently been highlighted.4
The aim of this study was to describe the main genotypic characteristics of a group of patients with LQTS and assess the usefulness of genetic testing in patients with IVF.
MethodsThe study included 9 patients who met the diagnostic criteria for LQTS (mean (SD) age, 22.6 (21.6) years; 66.7% women) and 4 patients with IVF (age, 26 (22.1) years; 50% women) who were assessed in our arrhythmias unit. The study included family history, laboratory workup, echocardiogram, and Holter monitoring. In the 4 patients with IVF and a normal corrected QT (QTc) interval, an electrophysiology study was carried out, along with coronary angiography, flecainide and epinephrine challenge. Genetic testing was carried out in 19 relatives of the probands with abnormal genotypes. All patients or their legal guardians provided signed informed consent.
A 5mL sample of peripheral blood was obtained for use as a sequencing template and the genes were sequenced by polymerase chain reaction. Only missense mutations that were not present in controls were considered pathologic.
ResultsThe clinical characteristics of the patients are shown in Table 1. The results of the clinical and genetic study are shown in Table 2. Of the 9 patients with LQTS, 7 (77.7%) had mutations: 71.4% in KCNH2 and 28.6% in SCN5A. No mutations were identified in KCNQ1. Only 2 of the mutations had previously been reported as associated with LQTS.2,5 Two patients (50%) with IVF had mutations, one in KCNH2 and the other in SCN5A; neither mutation had been described previously.
Table 1. Clinical Characteristics of Patients With Long QT Syndrome and Idiopathic Ventricular Fibrillation Included in the Study
| Sex | Age at first symptom | History of syncope | Age at diagnosis | Symptom leading to diagnosis | Phenotypic diagnosis | Cardiac arrest or ICD discharge | Triggers | |
| Patient 1 | Female | 11y | Yes | 49y | Cardiac arrest | LQTS | Yes | Noises |
| Patient 2 | Female | 60y | No | 67y | Cardiac arrest | LQTS | Yes | Emotions |
| Patient 3 | Female | 14y | Yes | 23y | Cardiac arrest | LQTS | Yes | Emotions |
| Patient 4 | Male | 17y | No | 24y | Cardiac arrest | IVF | Yes | Swimming |
| Patient 5 | Female | 10y | Yes | 11y | Cardiac arrest | IVF | Yes | Waking |
| Patient 6 | Male | 9y | Yes | 11y | Syncope | LQTS | No | Emotions |
| Patient 7 | Female | 53y | Yes | 58y | Cardiac arrest | IVF | Yes | Unknown |
| Patient 8 | Female | 0y | No | 1d | Cardiac arrest | LQTS | Yes | Rest |
| Patient 9 | Female | 19y | Yes | 20y | Syncope | LQTS | No | Rest |
| Patient 10 | Male | 2y | Yes | 10y | Syncope | LQTS | No | Unknown |
| Patient 11 | Female | 12y | Yes | 18y | Cardiac arrest | LQTS | Yes | Emotions |
| Patient 12 | Male | 10y | Yes | 11y | Syncope | IVF | Yes | Unknown |
| Patient 13 | Male | No | No | 5y | Asymptomatic | LQTS | No | No |
Abbreviations: d, days; ICD, implantable cardioverter defibrillator; IVF, idiopathic ventricular fibrillation; LQTS, long QT syndrome; y, years.
Table 2. Mutations Associated With Long QT Syndrome in Our Patient Series
| QTc | Pharmacological stress test (adrenaline/flecainide) | Phenotypic diagnosis | Diagnostic score | Treatment | Genetic study | Final diagnosis | |
| Patient 1 | 555 | No | LQTS | 8 | ICD | KCNH2+(1910 A>G Glu>Gly) | LQTS2 |
| Patient 2 | 478 | No | LQTS | 4 | ICD | SCN5A+(3985 G>R Gly>Ser) | LQTS3 |
| Patient 3 | 442 | + | LQTS | 4 | ICD | SCN5A + (1673 A>R His>Arg) | LQTS |
| Patient 4 | 367 | − | IVF | 1 | ICD | Negative | IVF |
| Patient 5 | 405 | − | IVF | 2 | ICD | KCNH2+(541 C>Y Arg>Trp) (577 G>R Al>Thr) (62 G>R Gly>Asp) | LQTS2 |
| Patient 6 | 631 | No | LQTS | 8 | ICD | KCNH2+(1714 G>R Gly> Ser) | LQTS2 |
| Patient 7 | 413 | − | IVF | 2 | ICD | SCN5A+(1673 A>R His>Arg) | IVF |
| Patient 8 | 720 | No | LQTS | 7.5 | ICD | Negative | LQTS |
| Patient 9 | 577 | No | LQTS | 4 | BB | Negative | LQTS |
| Patient 10 | − | No | LQTS | 4 | ICD | KCNH2+(343G>R Val>Met) | LQTS2 |
| Patient 11 | 488 | + | LQTS | 5 | ICD | KCNH2+(1882 G>S Gly>Arg) | LQTS2 |
| Paient 12 | 400 | − | IVF | 1 | ICD | Negative | IVF |
| Patient 13 | 476 | No | LQTS | 3.5 | No | KCNH2+(del 3079–3124) | LQTS2 |
Abbreviations: A, homozygous adenine; Al, alanine; Arg, arginine; Asp, aspartic acid; BB, beta blockers; C, homozygous cytosine; ICD, implantable cardioverter defibrillator; IVF, idiopathic ventricular fibrillation; G, homozygous guanine; Gly, glycine; Glu, glutamic acid; His, histidine; Met, methionine; R, heterozygous guanine/adenine; S, heterozygous guanine/cytosine; Ser, serine; LQTS, long QT syndrome; Thr, threonine; Trp, tryptophan; Val, valine.
Mutations were most commonly found in KCNH2. The mean age of the affected patients was 17.3 (16) years and the mean QTc interval was 511ms. We found 8 mutations (Table 2), only 2 of which had been previously described as associated with LQTS (G1882S and G1714R).2,5 Both are located between the P (pore) region and the fifth transmembrane domain, an essential region that acts as a selectivity filter for potassium.6
SCN5AOf the patients with LQTS in whom a mutation was identified, 28.6% had previously undescribed missense mutations in SCN5A. A female patient with IVF had a mutation for which there was no evidence of a direct causal relationship with LQTS. However, the clinical course supported a diagnosis of type 3 LQTS; there were implantable cardioverter defibrillator (ICD) discharges, with torsade de pointes episodes recorded, and episodes of paroxysmal atrial fibrillation that responded to flecainide challenge.
Diagnostic and Prognostic Value of Genetic Testing. Usefulness in Idiopathic Ventricular FibrillationAt the beginning of the study, there were 5 patients with no known heart disease and a normal QTc interval. The pharmacological stress test with adrenaline was normal in 4 of those patients and diagnostic of LQTS in 1 patient, who also had a positive result in genetic testing. In 2 of the 4 patients with IVF and a QTc interval at baseline and following adrenaline administration that was within the normal range, missense mutations were found during genetic testing.
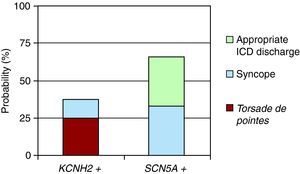
Figure 1 shows the events that occurred in patients treated with beta blockers according to the affected channel. Three patients with mutations in KCNH2 presented episodes: 2 cases of torsade de pointes polymorphic ventricular tachycardia and 1 case of syncope. Two episodes were recorded in patients with mutations in SCN5A: 1 episode of syncope and 1 inappropriate ICD discharge.

Figure 1. Probability of events (syncope, appropriate implantable cardioverter defibrillator [ICD] discharge, or torsade de pointes) following treatment with beta blockers (P>.05).
Genetic Testing in Asymptomatic Family MembersGenetic testing was carried out in 19 direct relatives of the patients with mutations. The mutation present in the index case was found in 6 relatives (31.5%); in 2 of those, the phenotype was negative (silent carriers).
In the 2 cases with previously described mutations, the 7 relatives who were tested did not carry the mutation. All had a normal QTc, and the disease was therefore ruled out.
DiscussionAlthough isolated mutations have been reported,7 no data are available from Spanish patient series addressing the genetic characteristics of LQTS. Our study provides preliminary data on the genotype profile of a small group of Spanish patients with LQTS and IVF. A multicenter study will be necessary to obtain larger groups and draw conclusions that can be extrapolated to the general population. Previous studies showed that genetic alterations are found in 65%–70% of cases and that the most frequently mutated gene is KCNQ1.1,8 We observed a slightly higher frequency of mutations (77.7%), and in our patients KCNH2 was the most commonly affected gene. These results cannot be extrapolated, however, given the small sample size.
Of the 9 missense mutations found, only 2 had previously been described as having a causal relationship with LQTS. Although the disease could be ruled out in asymptomatic relatives of these patients, analysis of the biophysical properties of the mutated ion channels would be necessary to unequivocally establish pathogenicity. However, data such as the region of the channel that is affected, the resulting amino acid change, and the absence of this protein in control subjects can help to assess the potential pathogenicity of the mutation.9
In our case series, 22.2% of the patients with LQTS and 50% of those with IVF had a negative genotype. This finding highlights the need to perform more extensive genetic testing in negative cases to include all other genes that have been described, along with the importance of exploring new candidate genes to explain the etiology of LQTS.10,11
The proportion of patients with arrhythmic events following treatment with beta blockers, which are more frequent in LQTS3 was high. This is explained mainly by selection bias due to patient recruitment in a hospital setting, with 8 out of 13 cases resuscitated following cardiac arrest.
Mutations were found in 2 of the 4 patients with IVF. Although we cannot assess the pathogenic impact of these mutations, one of the patients had 3 mutations in the N-terminal region of KCHN2; these mutations would have a high probability of altering the potassium current through the channel. Our study suggests that genetic testing has diagnostic value in patients who have survived ventricular fibrillation and in whom a full diagnostic workup has not yielded a diagnosis, even after negative pharmacological tests.
Of the relatives studied, 31.5% were carriers of the mutation and 33% of those were silent carriers. Although much remains to be understood about the prognosis of silent carriers, prohibition of drugs that lengthen the QT interval is essential and initiation of treatment with beta blockers should be considered.
In conclusion, our initial experience shows that genetic testing had a high level of sensitivity in a small series of patients with LQTS. Our observations that subtype 2 was the most prevalent differ from those of previous studies. Our study also suggests that genetic testing for mutations in these 3 genes is useful in IVF. Further studies in larger groups of patients will be necessary to confirm our findings.
Conflicts of InterestThe authors state that they have no conflicts of interest.
Received 22 January 2010
Accepted 25 February 2010
Corresponding author. Servicio de Cardiología, Hospital Universitario Virgen de las Nieves, Avda. de las Fuerzas Armadas 2, 18014 Granada, Spain. jimenez.jaimez@gmail.com