Hypertrophic cardiomyopathy (HCM) is diagnosed by the presence of ventricular hypertrophy ≥ 15 mm in the absence of any abnormal loading conditions that could cause it, or ≥ 13 mm if a relative is known to have HCM. On histological examination, myofibrillar disarray is characteristic of the disease. However, there have been reports of cases of HCM with sudden death (SD) in which the only identified abnormalities were myofibrillar disarray or mutations in the TNNT2 gene.1 We present a family with multiple cases of SD, in which pathological examination and genetic study were key in the diagnosis.
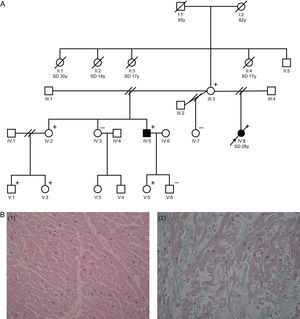
The proband was a 28-year-old woman, with no past medical history of note, who died suddenly while getting into her car. Postmortem examination revealed a heart weighing 295g with a 13-mm interventricular septum. Microscopic examination showed microscopic fiber hypertrophy and isolated foci of myofibrillar disarray (Figure 1B). Family history included 4 maternal aunts of the proband who had SD at the ages of 17 (n=2), 18, and 30 years (Figure 1A). The proband's 55-year-old mother had a normal echocardiogram and an electrocardiogram (ECG) with repolarization abnormalities in the inferolateral leads. The proband had 3 sisters and 1 brother, with different fathers. One of the sisters, aged 30 years, had a completely normal echocardiogram and cardiac magnetic resonance (CMR) scan, but ECG showed ST depression in the inferolateral leads. Stress echocardiography and coronary angiography showed no abnormalities. The other 2 sisters had normal echocardiograms and ECGs. In the brother, mild left ventricular hypertrophy was observed, with a 16-mm interventricular septum, and the ECG showed abnormalities similar to those of the sister and the mother (Table 1).
. B: histological images of the different cuts taken from the septum and left ventricle o f the proband (IV:8), showing the presence of a congestive cardiomyopathy, with regularly-organized hypertrophic myocytes (left image). Small areas can be seen on the free wall of the left ventricle where the fibers are disordered, lying obliquely and in swirls (disarray) (right image). SD, sudden death; y, years.")
A: family tree (square, male; circle, female; filled square or circle, phenotypically affected male/female; empty square or circle, healthy male or female; diagonal line, deceased; parallel diagonal lines, divorced). B: histological images of the different cuts taken from the septum and left ventricle o f the proband (IV:8), showing the presence of a congestive cardiomyopathy, with regularly-organized hypertrophic myocytes (left image). Small areas can be seen on the free wall of the left ventricle where the fibers are disordered, lying obliquely and in swirls (disarray) (right image). SD, sudden death; y, years.
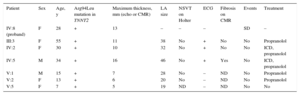
Clinical Characteristics of the Carriers of the Arg94Leu Mutation in TNNT2
| Patient | Sex | Age, y | Arg94Leu mutation in TNNT2 | Maximum thickness, mm (echo or CMR) | LA size | NSVT on Holter | ECG | Fibrosis on CMR | Events | Treatment |
|---|---|---|---|---|---|---|---|---|---|---|
| IV:8 (proband) | F | 28 | + | 13 | – | – | – | SD | – | |
| III:3 | F | 55 | + | 11 | 38 | No | + | No | No | Propranolol |
| IV:2 | F | 30 | + | 10 | 32 | No | + | No | No | ICD, propranolol |
| IV:5 | M | 34 | + | 16 | 46 | No | + | Yes | No | ICD, propranolol |
| V:1 | M | 15 | + | 7 | 28 | No | – | ND | No | Propranolol |
| V:2 | F | 13 | + | 6 | 20 | No | – | ND | No | Propranolol |
| V:5 | F | 7 | + | 5 | 19 | ND | – | ND | No | No |
CMR, cardiac magnetic resonance; ECG, electrocardiogram with repolarization abnormalities in the inferolateral leads; echo, echocardiography; F, female; ICD, implantable cardioverter-defibrillator; LA, left atrium; M, male; ND, not done; NSVT, nonsustained ventricular tachycardia.
Given the suspicion of familial HCM with mild phenotypic expression and the high incidence of SD, a genetic study was performed on a frozen blood sample from the deceased patient. We used a panel that identifies, through next-generation sequencing, multiple genes associated with cardiomyopathies and channelopathies, given the possibility of an underlying channelopathy in the deceased patients. An Arg94Leu mutation was identified in the troponin T gene (TNNT2). This mutation was first described in a British family with a high prevalence of premature SD (< 45 years old) and a diagnosis compatible with HCM. The patients had no history of illness, with SD being the first clinical manifestation. Postmortem examination revealed the absence of macroscopic hypertrophy, although histological examination showed diffuse fibrosis and myocyte disarray. In fact, that study was one of the first to establish that mutations in TNNT2 could be associated with SD even in the absence of overt hypertrophy.2,3 Two other mutations affecting the same residue have been described (Arg94Cys, Arg94His) and therefore it appears to be a point susceptible to mutations. This would suggest that any change in the amino acid sequence at this point would be poorly-tolerated. The clinical information available on carriers of these mutations agrees with other findings, such as the recently-published observations on the Arg92Gln mutation in several Mallorcan families.4 Some studies evaluating microscopic examination in carriers of troponin T mutations indicate that these cause less hypertrophy and fibrosis, but more disarray, than other sarcomere mutations. This could be the underlying factor that explains the high risk of arrhythmia.5
The genetic study of the rest of the family showed that the mother, 1 sister (and her 2 children), and the brother (and 1 of his daughters) were carriers of the identified mutation. The latest guidelines do not recommend automatic cardioverter-defibrillator implantation for the presence of a single mutation; however, a defibrillator was implanted as prevention in the proband's 2 carrier siblings, aged 33 and 35 years, due to patient preference (no risk factors except family history of SD) (Table 1). The other carriers, who are all under 16 except the proband's mother (55 years), are under close follow-up in clinic.
In conclusion, family history, pathological examination, and genetic study are key in the assessment of SD.6 The information from the family presented here indicates that HCM due to the Arg94Leu mutation in TNNT2 can present with ECG changes and histological disarray without macroscopic hypertrophy. The risk of SD in carriers is high and therefore risk should be carefully stratified.