
Left ventricular noncompaction (LVNC) is a structural abnormality characterized by a thin compacted epicardial layer and a thick endocardial layer with prominent trabeculations and deep recesses. When associated with cardiomyopathy, a genetic cause may be identified. Mutations in several genes, including tafazzin (TAZ), α-dystrobrevin (DTNA), Z-band alternatively spliced PDZ-motif protein (ZASP), lamin A/C (LMNA) and genes encoding sarcomeric proteins, have been reported.1 However, the yield of genetic testing appears to be relatively low, with clinically relevant variants identified in approximately 40% of cases. Frequently, limited information on the genes involved make it difficult to classify the variants identified.2 Most studies did not test for the presence of copy number variations (CNVs). CNVs, defined as gains or losses of DNA, contribute to the development of multiple genetic disorders and can be responsible for complex traits. In particular, 1p36 deletion has been associated with the development of LVNC cardiomyopathy in the context of a complex phenotype.3 Arndt et al.3 suggested that cardiac manifestations would likely depend on the involvement of the PRDM16 gene. Next-generation sequencing (NGS) coverage-based CNV analysis provides accurate tools for detecting CNVs. For this reason, the PRDM16 gene was included in our inherited cardiovascular diseases panels, and we systematically test our patients for CNVs in this gene. We identified 2 PRDM16 deletions in 382 patients referred with clinical suspicion of LVNC cardiomyopathy. No deletion affecting this gene was identified in more than 12 000 patients submitted with other cardiovascular phenotypes.
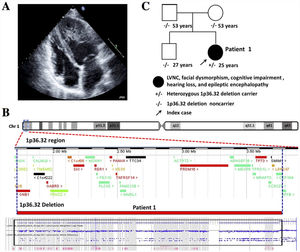
Patient 1 was a 25-year-old woman with LVNC cardiomyopathy (figure 1A) and complex phenotype diagnosed at 6 months old with mild facial dysmorphism, cognitive impairment, and epileptic encephalopathy. Sensorineural hearing loss was ruled out. Cardiac magnetic resonance imaging could not be performed due to claustrophobia and Holter monitoring showed no arrhythmias. Her parents and brother were unaffected. NGS coverage-based CNV analysis identified a heterozygous deletion of the PRDM16 gene. Single nucleotide polymorphism (SNP) array confirmed the deletion of a region comprising 112 genes, including PRDM16 (figure 1B). The deletion was confirmed to be de novo (figure 1C) and considered pathogenic.
. C: family segregation study by SNP array confirmed this deletion was de novo. CNVs, copy number variations.")
Patient 1. A: echocardiogram showing prominent medioapical left ventricular hypertrabeculation with moderate systolic dysfunction. B: CNV analysis by SNP array study confirmed the deletion of 112 genes including PRDM16 (Affymetrix CytoScan). C: family segregation study by SNP array confirmed this deletion was de novo. CNVs, copy number variations.
Patient 2 was a 23-year-old man evaluated after the sudden death of his father at age 43. In this case, autopsy was not performed. Echocardiogram showed LVNC without systolic impairment (figure 2A). Cardiac magnetic resonance imaging showed no late gadolinium enhancement and no arrhythmias were detected during Holter monitoring. No extracardiac anomalies were observed. NGS coverage-based CNV analysis identified a heterozygous deletion of PRDM16 exons 2 to 17 in the proband and also confirmed this deletion in his 20-year-old sister. She had LVNC and mild systolic dysfunction (figure 2B). SNP array in the proband confirmed a deletion of 11 genes, including PRDM16 (figure 2C). The deletion was absent in the unaffected mother (figure 2D) and considered likely pathogenic. However, the presence of the mutation could not be tested on the paternal side.
. D: family segregation study by NGS confirmed this deletion in the sister. CNVs, copy number variations; LVNC, left ventricular noncompaction.")
Patient 2. A: echocardiogram showing absence of compaction in the apical and lateral LV segments. B: LVNC in the affected sister. C: CNVs analysis by SNP array study confirmed the deletion of 11 genes including PRDM16 (Affymetrix CytoScan). D: family segregation study by NGS confirmed this deletion in the sister. CNVs, copy number variations; LVNC, left ventricular noncompaction.
Heterozygous 1p36 deletion is the most common subtelomeric deletion syndrome. It is believed to affect between 1 in 5000 and 1 in 10 000 newborns, although these may be underestimates. The associated phenotype is characterized by psychomotor retardation, hearing deficits, seizures, dysmorphic facial features, noncompaction/dilated cardiomyopathy, intellectual disability, and other congenital anomalies.4 There is high clinical variability among individuals, which could be explained by variation in the length of the deletions or the genes involved. Some of these genes have been individually associated with heart defects and/or developmental abnormalities.5 Arndt et al.3 used in situ hybridization to identify a common deletion region among patients with 1p36 deletion syndrome who presented with cardiomyopathy. This region included only the terminal 14 exons of PRDM16. These authors also found pathogenic variants in a proportion of nonsyndromic LVNC and dilated cardiomyopathy. Other chromosomal abnormalities (eg, 1q43 deletion, 5q35 deletion, and 8p23.1 deletion) have been reported in syndromic LVNC.6
We present 2 new cases of LVNC cardiomyopathy associated with 1p36 deletion syndrome in a large cohort of patients with suspicion of the disease, one of them with a nonsyndromic presentation, showing the usefulness of NGS in the identification of the genetic cause of the disease. Genetic testing with CNVs analysis should be included in the diagnostic protocol of LVNC cardiomyopathy. It could also lead to the identification of new candidate genes included in the CNV regions.
In conclusion, routine screening for CNVs in NGS-based genetic testing can be useful in the study of patients with LVNC cardiomyopathy. Complementary methods are usually required to confirm/characterize this type of variant. This strategy could increase the diagnostic yield in these patients, limiting the false negative rate. These data are of strong importance for adequate genetic counseling and screening of their families.
FUNDINGThe authors received no specific funding for this work
CONFLICTS OF INTERESTJ.P. Trujillo-Quintero, D. De Uña-Iglesias and L. Monserrat are members of the genetic diagnostics company Health in Code.