



The term inherited cardiovascular disease encompasses a group of cardiovascular diseases (cardiomyopathies, channelopathies, certain aortic diseases, and other syndromes) with a number of common characteristics: they have a genetic basis, a familial presentation, a heterogeneous clinical course, and, finally, can all be associated with sudden cardiac death. The present document summarizes some important concepts related to recent advances in sequencing techniques and understanding of the genetic bases of these diseases. We propose diagnostic algorithms and clinical practice recommendations and discuss controversial aspects of current clinical interest. We highlight the role of multidisciplinary referral units in the diagnosis and treatment of these conditions.
Keywords
The term inherited cardiovascular diseases (ICVD) covers a group of cardiovascular diseases (cardiomyopathies, channelopathies, certain aortic diseases, and other syndromes) with several common characteristics:
- 1.
They have a familial presentation. Thus, the relatives of affected people should be studied because they may have also inherited the disease. “When we treat a person with inherited cardiovascular disease, we do not just evaluate the patient, but the family as well”.1 Because families are studied, coordination with pediatric cardiology units is vital.
- 2.
They have a genetic basis and can currently be diagnosed using genetic techniques. Although not all of the causal mutations of the ICVD have been identified, the percentage of mutations identified has increased in recent years, reaching greater than 50% for some cardiomyopathies and channelopathies, such as hypertrophic cardiomyopathy (HCM), arrhythmogenic cardiomyopathy, and long QT syndrome (Table 1).
Table 1.Percentage of Patients Whose Inherited Cardiovascular Disease Can Be Linked to a Causal Mutation
Inherited cardiovascular disease Positive genetic studies*, % Hypertrophic cardiomyopathy 40-70 (Elliott et al2,3) Dilated cardiomyopathy 30 (Elliott et al3, Ackerman et al4) Restrictive cardiomyopathy Unknown (Elliott et al3, Ackerman et al4) Noncompaction cardiomyopathy 17-41 (Elliott et al3, Ackerman et al4) Arrhythmogenic dysplasia/cardiomyopathy 60 (Elliott et al3, Ackerman et al4) Brugada syndrome 20-30 (Ackerman et al4) CPVT 60-70 (Ackerman et al4) Long QT syndrome 70-80 (Ackerman et al4) Short QT syndrome Unknown (Ackerman et al4) Marfan syndrome 70-93 (Loeys et al5) Loeys-Dietz syndrome Depends on the clinical/imaging evaluation (Arslan-Kirchner et al6) CPVT, catecholaminergic polymorphic ventricular tachycardia.
- 3.
They can cause sudden cardiac death, sometimes as the first presentation of the disease. Sudden cardiac death has a high social, economic, and media impact. The leading cause of sudden cardiac death of elderly individuals with coronary heart disease risk factors is ischemic heart disease, whereas ICVDs are frequent causes of sudden death in those younger than 35 years (both athletes and nonathletes).7,8
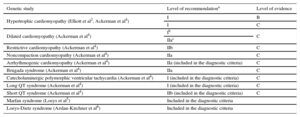
In recent years, various consensus documents and clinical guidelines have been published worldwide on the diagnosis and treatment of cardiomyopathies, channelopathies, and various aortic diseases with genetic origins. All agree on the need (with different recommendation and evidence levels) to study the relatives of affected patients, as well as to perform genetic studies (Table 2).
Recommendations and Level of Evidence of Genetic Studies in the Guidelines and Published Consensus Documents
| Genetic study | Level of recommendationa | Level of evidence |
|---|---|---|
| Hypertrophic cardiomyopathy (Elliott et al2, Ackerman et al4) | I | B |
| I | C | |
| Dilated cardiomyopathy (Ackerman et al4) | Ib | C |
| IIac | ||
| Restrictive cardiomyopathy (Ackerman et al4) | IIb | C |
| Noncompaction cardiomyopathy (Ackerman et al4) | IIa | C |
| Arrhythmogenic cardiomyopathy (Ackerman et al4) | IIa (included in the diagnostic criteria) | C |
| Brugada syndrome (Ackerman et al4) | IIa | C |
| Catecholaminergic polymorphic ventricular tachycardia (Ackerman et al4) | I (included in the diagnostic criteria) | C |
| Long QT syndrome (Ackerman et al4) | I (included in the diagnostic criteria) | C |
| Short QT syndrome (Ackerman et al4) | IIb (included in the diagnostic criteria) | C |
| Marfan syndrome (Loeys et al5) | Included in the diagnostic criteria | |
| Loeys-Dietz syndrome (Arslan-Kirchner et al6) | Included in the diagnostic criteria |
I, recommended; IIa, can be useful; IIb, may be considered.
To diagnose and treat ICVDs, we also highlight here the creation of specialized referral units. These units should be multidisciplinary and maintain close links with other services and specialities.2,9 However, in Spain, these referral units are unavailable in some communities, making it sometimes difficult to comply with many of the recommendations of these documents.
The present document has been created by the Working Group on Inherited Cardiovascular Disease of the Spanish Society of Cardiology (Sociedad Española de Cardiología [SEC]) in collaboration with the Working Group on the Aorta (from the Clinical Cardiology Section), the Working Group on Sports Cardiology, the Electrophysiology and Arrhythmias Section, the Heart Failure and Heart Transplant Section of the SEC, and the Spanish Society of Pediatric Cardiology and Congenital Heart Diseases. Moreover, the document has been reviewed by a group of Spanish and international experts (Appendix).
One of the aims of this document was to explain various aspects of the care of patients with ICVD in order to bridge the gap between Spanish clinical practice and the guidelines and recommendations.
The Spanish Ministry of Health has designated several referral units (Centros, Servicios y Unidades de Referencia in the Spanish National Health System) for the diagnosis and treatment of ICVD.10 These referral units are essential for complex cases, namely, those with diagnostic and/or treatment difficulties, but we believe that the “way of working” of the ICVD units should be exported to all health care professionals (eg, cardiologists, internists, cardiovascular surgeons, geneticists, pediatricians, nurses, psychologists) who may be involved in the study and treatment of these conditions.
Additionally, we have paid special attention to certain aspects and terms due to their novelty or multidisciplinary characteristics. Finally, certain basic recommendations have been highlighted (Table 3), to be used as jumping-off points to further the understanding and treatment of these diseases.
Recommendations for the Management of Inherited Cardiovascular Diseases
| I | A family tree of at least 3 generations should be drawn and each member should be investigated for possible diseases related to the ICVD being studied. The effort to recall the symptoms and signs of relatives not only sometimes allows the detection of affected or possibly affected family members, but also allows determination of the inheritance pattern |
| II | When treating patients with ICVD, evaluation of relatives is necessary. A specialist or unit with experience should evaluate the family members and integrate the results of all of the relatives. Follow-up should be provided to relatives at risk of the disease. The family study should be facilitated by the genetic study when a pathogenic mutation has been identified in the family |
| III | Genetic studies should be included in the standard clinical diagnostic arsenal of ICVD. Their indications depend on their diagnostic yield for the disease (proportion of positive results) and the value of their results in the diagnosis and prognosis of the affected individuals and their relatives |
| IV | When treating patients with ICVD, heart disease caused by a phenocopy should be specifically ruled out, because these conditions usually have a different clinical course and treatment |
| V | The clinical study of the relatives of patients with ICVD should be begun independently of age. For the genetic study of relatives of patients with cardiomyopathies or hereditary aortic diseases, the recommend starting age is 10 years (at diagnosis in the case of channelopathies). This age is orientative and depends on the severity of the familial phenotype and the particularities of each family and each study center |
| VI | Reproductive counseling should be given to patients desiring to have children. Genetic counselors should be associated with units with expertise on pathology, legislation, and reproductive techniques. Spanish law should make an effort to adapt to the new reality of ICVD and we propose the creation of working groups to develop this field and facilitate this task |
| VII | Blood and/or tissue samples should be collected from those who die of sudden cardiac death with suspected or confirmed ICVD and stored to permit a genetic study (“molecular autopsy”). Each autonomous community in Spain should provide the means to create referral centers for the macroscopic and microscopic study of the hearts and aortas of young patients with suspected ICVD who die suddenly. These centers should coordinate with ICVD units. At the same time, stable cooperative ties should be established between the justice (in charge of coroners and forensic pathologists) and health (in charge of ICVD units) administrations |
| VIII | Psychologists or personnel specialized in psychological support should be incorporated into ICVD units |
| IX | Patients with ICVD should be encouraged and helped to create patient associations that can provide additional support not only to affected people, but also to those healthy family members living with them |
ICVD, inherited cardiovascular disease.
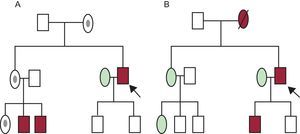
A family tree, genealogical tree, or medical pedigree is a graphical representation of the medical history and kinship of a family. With recent advances in genetic techniques and their increasingly widespread use, all physicians (or health care professionals) should be able to draw and interpret a family tree.
A detailed family tree with 3 generations should be created as part of the evaluation of patients with ICVD (Figure 1). Questions should be formulated to extract the relevant information on the cardiological history and symptoms suggesting a cardiac origin.11,12

Family trees or pedigrees of 2 families with an inherited cardiovascular disease. A: X-linked transmission. B: autosomal dominant transmission. Rectangle, male; oval, female; red rectangle or oval, male or female phenotypically affected; rectangle or oval without frame, healthy male or female; dark circle inside the oval, healthy carrier; arrow, proband or index patient; symbol with diagonal line, deceased person.
Thus, it is important: a) to identify the index patient (or proband), that is, the first case evaluated in the family (can be alive or dead); b) record names, birthdates, and causes and ages of deaths (including sudden infant death), while always respecting the applicable data protection laws; c) to rule out the existence of consanguinity and to ask about the geographical origins of the family; d) to investigate symptoms, signs, complications, and treatments (eg, defibrillators, pacemakers, heart/kidney transplant) potentially related to the ICVD being studied, and e) to include all available documentation in the family study, including clinical history and previous electrocardiogram results. It is also crucial to locate any autopsy reports.
TYPES OF INHERITANCEMost ICVD have an autosomal dominant mode of transmission, that is, a patient can transmit the disease to both males and females, and their offspring have a 50% chance of inheriting the genetic defect causing the disease. Autosomal dominant inheritance is confirmed if a “male-to-male disease transmission” is seen within the family tree.
When there is history of consanguinity, autosomal recessive inheritance is more likely. In this situation, each descendent has a 25% chance of inheriting 2 mutated alleles. Female-to-male transmission with a healthy mother and affected father is highly indicative of recessive X-linked disease. Female-to-female or -male transmission without transmission from the male to the offspring indicates matrilineal inheritance (typical of mitochondrial diseases). A mutation can sometimes appear de novo in the index patient, meaning that although the patient's parents do not have the mutation, it can be transmitted to any descendents.11,13
Recommendation I: a family tree of at least 3 generations should be drawn and each member should be investigated for possible diseases related to the ICVD being studied. The effort to recall the symptoms and signs of relatives not only sometimes allows the detection of affected or possibly affected family members, but also allows determination of the inheritance pattern.
DIAGNOSTIC CRITERIA: FOR INDEX PATIENTS AND RELATIVESThe consensus documents and guidelines describing the diagnostic criteria of the main ICVD are shown in Table 4.
Consensus Documents and Guidelines Describing the Diagnostic Criteria of the Main Inherited Cardiovascular Diseases
| Inherited cardiovascular disease | Document |
|---|---|
| Hypertrophic cardiomyopathy | 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy. The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (Elliott et al2) Comments on the clinical practice guidelines of the European Society of Cardiology 2014 on the diagnosis and management of hypertrophic cardiomyopathy. A critical outlook from the point of view of Spanish cardiology (Working Group of the SEC14) |
| Cardiomyopathies | Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases (Charron et al11) |
| Channelopathies | HRS/EHRA/APHRS Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromes (Priori et al9) |
| Dilated cardiomyopathy* | Guidelines for the study of familial dilated cardiomyopathies. Collaborative Research Group of the European Human and Capital Mobility Project on Familial Dilated Cardiomyopathy (Mestroni et al15) |
| Cardiomyopathies and channelopathies | HRS/EHRA Expert Consensus Statement on the State of Genetic Testing for the Channelopathies and Cardiomyopathies (Ackerman et al4) |
| Cardiomyopathies | Genetic Evaluation of Cardiomyopathy: A Heart Failure Society of America Practice Guideline (Hershberger et al16) |
| Arrhythmogenic cardiomyopathy | Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria (Marcus et al17) |
| Thoracic aortic disease | 2010 CCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with Thoracic Aortic Disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine (Hiratzka et al18) |
| Marfan syndrome | The revised Ghent nosology for the Marfan syndrome (Loeys et al5) |
| Aortic disease | 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases: Document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (Erbel et al19) Comments on the clinical practice guidelines of the SEC 2014 on the diagnosis and treatment of aortic diseases (Working Group of the SEC20) |
AATS, American Association for Thoracic Surgery; ACCF, American College of Cardiology Foundation; ACR, American College of Radiology; AHA, American Heart Association; APHRS, Asia Pacific Heart Rhythm Society; ASA, American Stroke Association; EHRA, European Heart Rhythm Association; ESC, European Society of Cardiology; HRS, Heart Rhythm Society; SCA, Society of Cardiovascular Anesthesiologists; SCAI, Society for Cardiovascular Angiography and Interventions; SEC, Spanish Society of Cardiology (Sociedad Española de Cardiología); SIR, Society of Interventional Radiology; STS, Society of Thoracic Surgeons; SVM, Society for Vascular Medicine.
In some ICVD a distinction is made between the diagnostic criteria applied to index patients and the criteria—generally less stringent—applied to relatives, which ultimately facilitate the earlier diagnosis of the disease. Early diagnosis in family members may involve life style changes and treatment initiation and can positively impact disease prognosis.
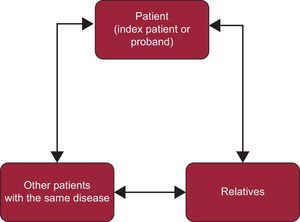
PROTOCOLS AND DIAGNOSTIC ALGORITHMSIn ICVDs not only the index patients or probands (definitely affected) should be studied, but also their relatives (who may or may not be affected). The information obtained from index patients is vital for the investigation of relatives. Similarly, the information from family members can be fundamental for a definitive diagnosis of index patients (Figure 2).

For the diagnosis and treatment of uncommon diseases such as ICVD both local and national or international registries are vital.
Next, general diagnostic algorithms are proposed for the 2 most common situations in ICVD consultations:
- 1.
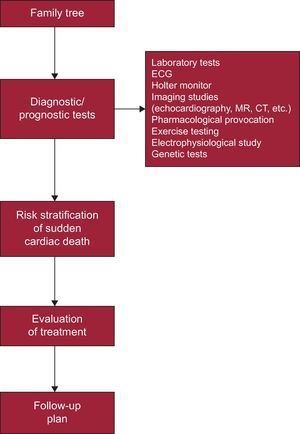
Patient with suspected ICVD (eg, cardiomyopathy, channelopathy, aortic disease) (Figure 3).
- 2.
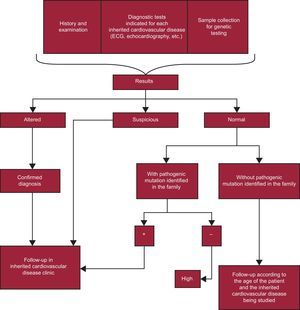
Relative of patient with a diagnosed ICVD (Figure 4).
, and due to the possibility that there is more than one mutation in the family (eg, an affected family member could be the carrier of an unidentified mutation in the genetic study performed). This approach should be applied to the family studies of the different heart diseases. ECG, electrocardiogram.") Figure 4.
Figure 4.Action algorithm for families of patients diagnosed with an inherited cardiovascular disease. According to the recommendations of the guidelines for the diagnosis and treatment of hypertrophic cardiomyopathy of the European Society of Cardiology of 2014, patients can be discharged after a clinical evaluation involving echocardiography and electrocardiography if they are not carriers of a familial mutation. The Working Group on Inherited Cardiovascular Diseases of the Spanish Society of Cardiology believes that no relatives of a patient with cardiomyopathy should be discharged without undergoing echocardiography and electrocardiography. These tests are vital due to their usefulness in establishing the cosegregation of the mutation in each family, which supports its pathogenicity (often for de novo mutations, without previous reports in the literature), and due to the possibility that there is more than one mutation in the family (eg, an affected family member could be the carrier of an unidentified mutation in the genetic study performed). This approach should be applied to the family studies of the different heart diseases. ECG, electrocardiogram.
(0.34MB).

, and due to the possibility that there is more than one mutation in the family (eg, an affected family member could be the carrier of an unidentified mutation in the genetic study performed). This approach should be applied to the family studies of the different heart diseases. ECG, electrocardiogram.")
To complete the diagnosis and establish the prognosis of the family, the physician in charge from the clinic or ICVD unit should integrate all of the information gathered from the 2 parts.
Recommendation II: when treating patients with ICVD evaluation of relatives is necessary. A specialist or unit with experience should evaluate the family members and integrate the results of all of the relatives. Follow-up should be provided to relatives at risk of the disease. The family study should be facilitated by the genetic study when a pathogenic mutation has been identified in the family.
GENETICSGenetic studies have radically changed our approach to ICVD and are now a fundamental tool in diagnostic algorithms. Until recently, genetic studies were limited to a few genes and were highly expensive (Sanger method). Currently, due to the development of massive sequencing techniques (next-generation sequencing [NGS]), hundreds of genes can be studied quickly and at reasonable prices, and even the entire genome (or the coding region, known as the exome). Because so many genes can be studied, the main problem with NGS is that it can be difficult to evaluate the pathogenicity of multiple variants.21
All of the recent guidelines and consensus documents on ICVD already confer genetic studies with the highest recommendation levels (Table 2).
Genetic studies of ICVD have a diagnostic utility and can be directly applied to facilitate reproductive/professional advice and schedule follow-up of families. In some cases, the genetic study also has implications for the treatment and prognosis of the patients themselves.4,22,23
Recommendation III: genetic studies should be included in the standard clinical diagnostic arsenal of ICVD. Their indications depend on their diagnostic yield for the disease (proportion of positive results) and the value of their results in the diagnosis and prognosis of the affected individuals and their relatives.
PHENOCOPIESThe term phenocopy is used to describe patients who have the same phenotype (characteristics features of a disease) as other individuals with a known genetic alteration but who do not have the same mutation.3 There are phenocopies of cardiomyopathies as well as channelopathies and aortic diseases.
We believe that this concept is unclear. For example, “ventricular hypertrophic diseases”, produced by mutations in the GLA gene (Fabry disease) or in LAMP2 (Danon disease), among many others, are considered phenocopies of HCM. However, according to the 2008 European classification of cardiomyopathies,3 which are grouped according to morphology and ventricular function, all of these conditions are HCM. For HCM, the term phenocopy is based on considering only those hypertrophies caused by mutations in sarcomeric genes to be HCM. Strictly speaking, the term phenocopy could be used for hypertrophy due to “athlete's heart”.24
Another example is catecholaminergic polymorphic ventricular tachycardia, a condition diagnosed by a structurally normal heart with normal electrocardiogram and polymorphic ventricular arrhythmias during exercise testing or adrenaline stimulation. Generally, catecholaminergic polymorphic ventricular tachycardia is caused by mutations in RYR2 and CASQ2. However, mutations in other genes related to distinct types of Another example is catecholaminergic can also produce arrhythmias resembling the classic description of catecholaminergic polymorphic ventricular tachycardia, such as in Andersen-Tawil syndrome (due to mutations in KCNJ2 and long QT syndrome type 7) or mutations in ANK2 (long QT syndrome type 4).4
The same situation can occur in Marfan syndrome, caused by mutations in the fibrillin 1 FB1 (fibrillin 1) gene. Some years ago, 2 types of Marfan syndrome were distinguished: type 1, caused by mutations in FBN1, and type 2, caused by mutations in TGFBR1 and TGFBR2. These patient subtypes were subsequently expanded to include patients with marfanoid characteristics but with aggressive vascular disease and other characteristic morphologies, such as hypertelorism, bifid uvula, and tortuous artery in Loeys-Dietz syndrome.13
Familiarity with these “phenocopies” is important because some can have a more aggressive behavior than the disease they “imitate” and because many have specific treatments (eg, enzyme replacement therapy in Fabry disease) or require more aggressive therapeutic measures (eg, earlier surgery in Loeys-Dietz syndrome). Thus, the diagnostic protocols in ICVD should include imaging and/or laboratory tests to rule out possible phenocopies.
OVERLAPPING PHENOTYPESThe following cardiomyopathy phenotypes can be found: HCM, restrictive cardiomyopathy, dilated cardiomyopathy, arrhythmogenic cardiomyopathy, and noncompaction cardiomyopathy.4 These conditions have classically been considered distinct and independent diseases but it is now recognized that patients can move from one phenotype to another (eg, patient with HCM shifting to dilated cardiomyopathy) or that various phenotypes in the same family can be associated with the same mutation (eg, patient with noncompaction cardiomyopathy and relatives with HCM). A similar phenomenon occurs in channelopathies. For example, mutations in SCN5A can cause either long QT syndrome or Brugada syndrome.25
Genetics plays a fundamental role in the understanding of these diseases. These “overlapping phenotypes” may be due to mutations in different regions of the same gene having different effects, but another possibility is that the disease is the same, even though it shows a different phenotype.
Recommendation IV: when treating patients with ICVD heart disease caused by a phenocopy should be specifically ruled out, because these conditions usually have a different clinical course and treatment.
INTERPRETATION OF GENETIC STUDIESBasically, the following are considered25:
- 1.
Classification by frequency:
- a.
Polymorphism: involves a change in the nucleotide sequence that is also found in the general healthy population (in at least 0.5%-1.0%).
- b.
Rare variants: changes found in a very low percentage of the general healthy population (< 0.5%).
- c.
Mutations: changes not found in the healthy population.
- a.
- 2.
Classification by pathogenicity: monogenic diseases, such as most ICVD, are caused by mutations or rare variants; most polymorphisms are not pathogenic but certain polymorphisms can sometimes act as modulators or modifiers that cause disease in the presence of certain environmental factors (eg, drugs). Thus:
- a.
The presence of a pathogenic variant (usually a mutation) is used to confirm the diagnosis in the index patient, complete the study and follow-up of related carriers, and sometimes indicate the prognosis and enable the discharge of related noncarriers from the clinic.
- b.
The identification of a benign variant (typically a polymorphism) in the index patient does not help to confirm the diagnosis or the family study, except when the variant can act as a modifier or modulator.
- c.
The VUS (variant of unknown significance), or unknown pathogenicity. These variants involve changes in the nucleotide sequence with unknown disease effects according to current understanding. Thus, VUSs cannot confirm the diagnosis or be used in the family study. Some of these variants can be studied in the family but for research purposes. In a recent consensus document, the European Society of Cardiology recommended maintaining contact with carriers of VUSs in case their pathogenicity is later established.12,21
- a.
Thus, the genetic study of a ICVD sometimes fails to detect the pathogenic mutation causing the disease. The proportion of positive results of the genetic study varies between 20% and 90%, depending on the disease (Table 1). The lack of an identifiable pathogenic mutation in a family diagnosed by clinical ICVD criteria does not mean that they do not have the disease. Because not all genes involved in the ICVD have been identified, it must be considered that the disease affects the family and the relatives should be evaluated in accordance with the clinical criteria.
WHEN SHOULD WE START A CLINICAL AND GENETIC STUDY OF RELATIVES OF PATIENTS WITH INHERITED CARDIOVASCULAR DISEASE?This is a highly controversial topic. The consensus documents and guidelines sometimes make somewhat vague recommendations, leaving the final decision to the physicians and families. The European Society of Human Genetics recommend delaying the genetic studies in children, arguing that the diagnosis can cause unjustifiable anxiety, as well as overprotective attitudes.26 On the other hand, many cardiologists (including pediatric cardiologists) argue that an uncertain ICVD status would cause greater anxiety than the knowledge of whether one is a mutation carrier or even affected.
Because the most frequent pattern of inheritance is autosomal dominant 50% of the offspring will not inherit the genetic defect and will not require periodic revisions, physical restrictions, or drugs (in the case of channelopathies).
A genetic study before the recommended age can be justified by a sudden cardiac death at an early age in the family, the desire to participate in sport activities or begin competitive sports, or the overwhelming need of a family to know whether their child has the familial mutation.2,4,6,11,13 Thus, the debate about when to perform the genetic studies continues.
We believe that family members should undergo a clinical evaluation regardless of their age, both for cardiomyopathies and channelopathies or aortic diseases. After the initial evaluation, the relatives should continue to undergo periodic follow-ups, depending on the disease and the severity of the phenotype.
However, for the genetic study, we should clearly distinguish between structural disease (cardiomyopathies and genetic diseases of the aorta) and nonstructural disease (channelopathies). First, the risk of complications is usually linked to the type and severity of the phenotype. Thus, if a conservative approach is chosen, some patients can wait until adulthood before undergoing a genetic study because, in the absence of phenotypic expression, the therapeutic approach is going to change little, if at all. On the other hand, an “interventionist” attitude and early genetic diagnosis are also possible as long as the results are correctly interpreted by the medical team and properly explained to the family.
Conversely, with channelopathies, early genetic studies are considered more apt, as the findings can trigger life style changes aimed at preventing major arrhythmic complications.
Recommendation V: the clinical study of the relatives of patients with ICVD should be begun independently of age.
For the genetic study of relatives of patients with cardiomyopathies or hereditary aortic diseases, the recommend starting age is 10 years (or at diagnosis in the case of channelopathies). This age is orientative and depends on the severity of the familial phenotype and the particularities of each family and each study center.
REPRODUCTIVE COUNSELINGOne of the consequences of genetic studies of ICVD is the implications for reproduction. The aim should be that patients know the possibility of transmitting the genetic alteration to their offspring, the implications of this transmission, and the available reproductive alternatives.
Many couples seek advice on the probability of having offspring who are free of the congenital disease. There are various ways to avoid transmission of a genetic defect, but all are subject to important ethical and legal aspects that should be born in mind:
- 1.
Prenatal diagnosis analyzes DNA extracted from fetal cells obtained from the chorionic villi (between weeks 10 and 12) or amniotic fluid cells (between weeks 14 and 16 of gestation). This analysis can be performed if the causal mutation in the family has already been identified, as long as the mutation has a clearly demonstrated pathogenicity, if disability is expected, or if there is a high risk of early death or there are no available effective treatments. Because prenatal diagnosis techniques have a risk of miscarriage, they are only justified if the pregnancy would be terminated if the fetus is determined to be a carrier of the genetic defect.
- 2.
Preimplantation diagnosis is performed in association with in vitro fertilization. This technique can reveal whether the in vitro fertilized embryos are carriers of the pathogenic mutation to ensure that implantation is limited to embryos free of the mutation. Spanish legislation allows these techniques to be used to detect severe, incurable, early-onset hereditary diseases.27 However, the absence of an explicit list of the diseases examinable using these techniques under this law is a source of confusion. These approaches are fully accepted for some ICVD such as Marfan disease and Fabry disease. For other ICVD individual authorization is required from the Spanish National Commission for Assisted Reproduction.13,27
- 3.
Other possibilities to preclude the transmission of the familial genetic defect are the use of donor eggs or sperm (depending on the affected progenitor) and adoption.
Recommendation VI: reproductive counseling should be given to patients desiring to have children. Genetic counselors should be associated with units with expertise on pathology, legislation, and reproductive techniques. Spanish law should make an effort to adapt to the new reality of ICVD and we propose the creation of working groups to develop this field and facilitate this task.
STUDY OF SUDDEN CARDIAC DEATHSudden cardiac death can be due to ICVD especially when it occurs in a young person (younger than 40 years).7,8 The correct diagnosis of the cause of death can avoid another fatal outcome in other members of the family.
After the sudden death of a young person, a complete autopsy should be performed according to specific protocols to determine the underlying cause of death.28–30 This autopsy should pay special attention to the heart and aorta and should be performed by forensic and/or pathology experts in ICVD. Even in the hands of experts, the cause of death can be elusive in a variable proportion of autopsies (between 4% and 50% of medicolegal autopsies; the higher percentages correspond to forensic series of children and adolescents30–32).
The tissue samples of the deceased should be preserved so that the case can be pathologically revised using different techniques and stains. For molecular and genetic studies, blood and tissue samples should be kept (without fixation). A genetic study, known as a “molecular autopsy”, can diagnose the cause of death in more than 35% of cases with a negative autopsy.33
Regardless of whether a definite diagnosis is obtained from the autopsy and, after ruling out a noninherited cardiovascular disease (largely ischemic or valvular), a cardiological study of the first-degree relatives is recommended (its extent depends on the suspected disease).
The expert documents and the guidelines9,34 specifically recommend the development of ICVD (or “familial arrhythmia”) clinics to study the causes of sudden cardiac death.
Recommendation VII: blood and/or tissue samples should be collected from those who die of sudden cardiac death with suspected or confirmed ICVD and stored to permit a genetic study (“molecular autopsy”). Each autonomous community in Spain should provide the means to create referral centers for the macroscopic and microscopic study of the hearts and aortas of young patients with suspected ICVD who die suddenly. These centers should coordinate with ICVD units. At the same time, stable cooperative ties should be established between the justice (in charge of coroners and forensic pathologists) and health (in charge of ICVD units) administrations.
PATIENT AND FAMILIAL SUPPORTAlthough the vast majority of patients with an ICVD can live a normal life, a small percentage has significant impairments or premature death. It is difficult to deal with the sudden loss of a family member, particularly when the deceased is a child or other youth.
Although there are some exceptions, the Spanish public health system has no specialized psychological units for these situations. Other medical fields, such as oncology, have extensive experience with psychological support programs.
In other European countries, such as the United Kingdom,35,36 patient associations perform a commendable job in supporting, monitoring, training (eg, in basic cardiopulmonary resuscitation techniques), and educating these patients. Although there are some similar associations in Spain,37,38 they are insufficient and lack this associative aspect that is so beneficial to patients. The information given by physicians to patients is important but no one can explain the feelings and experiences of having a disease as well as another person affected by the disease.
Recommendation VIII: psychologists or personnel specialized in psychological support should be incorporated into ICVD units.
Recommendation IX: patients with ICVD should be encouraged and helped to create patient associations that can provide additional support not only to affected people, but also to those healthy family members living with them.
CONCLUSIONSThe aim of this document was to export the “way of working” of the ICVD units to all health care professionals (eg, cardiologists, internists, cardiovascular surgeons, geneticists, pediatricians, nurses, psychologists) who may be involved in ICVD study and treatment. We have proposed diagnostic algorithms, discussed current controversial aspects of clinical interest, and set forth 9 basic recommendations. These recommendations should be considered a starting point to improve the understanding and treatment of these diseases. In addition to its doubtless value as a synthesis document of the current status of ICVD management, this work aimed to help to homogenize the process of care of these patients in Spain, which will undoubtedly improve the quality of the health care.
FUNDINGThis work has been partially financed by the Red de Investigación Cardiovascular of the Instituto de Salud Carlos III (RD12/0042/0069, RD12/0042/0049, RD12/0042/0029, RD12/0042/0069, RD12/0042/0021, RD12/0042/0021 RD12/0042/0066) and the European Regional Development Fund (ERDF, Una manera de hacer Europa).
CONFLICTS OF INTERESTNone declared.
| María Teresa Tomé-Esteban; St. George's, University of London, London, United Kingdom |
| Andrea Mazzanti; Fondazione Salvatore Maugeri, Pavia, Italy |
| Luis Rocha-Lopes; Hospital García de Orta, Almada, Portugal |
| Adrián Fernández; Hospital Universitario Fundación Favaloro, Buenos Aires, Argentina |
| Ricardo Stein; Universidade Federal do Rio Grande do Sul, Cardiología, Porto Alegre, Brazil |
| Ivonne J. Cárdenas-Reyes; Universidad del Rosario, Bogotá, Colombia |
| Alicia Beatriz Aguilera-Tapia; Instituto Nacional de Toxicología y Ciencias Forenses, Madrid, Spain |
| María Paz Suárez-Mier; Instituto Nacional de Toxicología y Ciencias Forenses, Madrid, Spain |
| Lorenzo Monserrat-Iglesias; Instituto de Investigación Biomédica de A Coruña, A Coruña, Spain |