
We present the case of a family with a history of sudden cardiac death at a young age. The initial diagnosis was catecholaminergic polymorphic ventricular tachycardia (CPVT) caused by the p.L3778F mutation in the cardiac ryanodine receptor gene (RyR2). Several years after this initial diagnosis, a comprehensive family genetic study by next generation sequencing (NGS) has now identified a second pathogenic mutation, this time in KCNQ1 and linked to type 1 long QT syndrome (LQTS1). The severe phenotype is likely due to the presence of both mutations and not exclusively to the p.L3778F mutation in RyR2 as previously reported.1
The proband (III:5) had a cardiac arrest at the age of 8 years while swimming but recovered fully after resuscitation. His brother had died suddenly, also while swimming, at 10 years of age (no abnormalities were detected on autopsy).
An electrocardiogram (ECG) of the proband showed sinus bradycardia with a QTc of 440ms, whereas the results of echocardiography, Holter recording, and exercise stress testing were normal. The context of the cardiac arrest suggested LQTS1, and the patient was prescribed beta-blockers. An electrophysiological study was conducted without arrhythmia induction, and the patient was fitted with a subcutaneous Holter monitor. At the age of 10 years, he had an exertion-induced syncopal episode, and ECG revealed self-limiting polymorphic ventricular tachycardia (Figure A).
 or the E449R*14 mutation in KCNQ1 (red); +, carriers, heterozygous for L3778F in RyR2 (blue) or E449R*14 in KCNQ1 (red); circles, women; boxes, men; arrow, proband; blue fill, patient with CPVT (according to the European and US consensus statement)3; black fill, patient clinically affected by LQTS1 and CPVT; red fill, patient with LQTS1; RyR2, ryanodine receptor gene; LQTS1, type 1 long QT syndrome; CPVT, catecholaminergic polymorphic ventricular tachycardia.")
A: Recording of the polymorphic tachycardia in the proband. B: Family tree./, deceased; –, noncarriers, lacking the L3778F mutation in RyR2 (blue) or the E449R*14 mutation in KCNQ1 (red); +, carriers, heterozygous for L3778F in RyR2 (blue) or E449R*14 in KCNQ1 (red); circles, women; boxes, men; arrow, proband; blue fill, patient with CPVT (according to the European and US consensus statement)3; black fill, patient clinically affected by LQTS1 and CPVT; red fill, patient with LQTS1; RyR2, ryanodine receptor gene; LQTS1, type 1 long QT syndrome; CPVT, catecholaminergic polymorphic ventricular tachycardia.
The patient was referred for further investigation to a center with expertise in cardiac channelopathies. A mutational screen was conducted of the RyR2, KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2 genes by denaturing high performance liquid chromatography (DHPLC) and Sanger sequencing, identifying a heterozygous missense mutation in RyR2 (p.L3778F). Beta-blockers were prescribed, and the patient underwent left sympathetic denervation and was fitted with an implantable cardioverter-defibrillator. The patient's immediate family members were asymptomatic, but the father and sister were identified as carriers of the mutation (Figure B). The mutation had not been described before and the case was included in a study of CPVT patients that highlighted the poor prognosis of male carriers.1
Several years later, the family relocated and the proband's 5-year-old niece (IV:1) was referred for evaluation. She was asymptomatic, but the study confirmed that she carried the family mutation. Interestingly, her 63-year-old grandfather (II:7) and 39-year-old mother (III:6) were both carriers and yet were asymptomatic on ECG, exercise stress testing, and Holter recording (except for an exercise stress test in the grandfather that showed an isolated premature ventricular contraction in 2 morphologies upon exercise initiation, which disappeared with exertion).
Suspicion of a second mutation prompted a new genetic study of the proband by NGS (195 genes). In addition to the p.L3778F mutation in RyR2, a second mutation was detected, in KCNQ1 (g.2610034_2610035insC); this mutation generates an aberrant transcript with a stop codon in amino acid position 463 (p.E449R*14). The mutation had been previously reported in relation to LQTS1,2 but was not detected in the original genetic analysis by DHPLC and Sanger sequencing.1
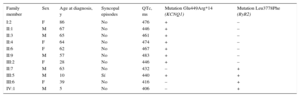
Further analysis in partnership with other centers confirmed the KCNQ1 mutation in 7 family members (Figure B). All affected individuals were asymptomatic, yet the mean QTc on basal ECG was 460 ± 15ms (Table). Given that 25% of LQTS1 patients can have a normal QTc on basal ECG, the carriers of this mutation were diagnosed with LQTS1, according to current diagnostic criteria.3
Characteristics of Carriers of the Mutations in KCNQ1 or RyR2
| Family member | Sex | Age at diagnosis, y | Syncopal episodes | QTc, ms | Mutation Glu449Arg*14 (KCNQ1) | Mutation Leu3778Phe (RyR2) |
|---|---|---|---|---|---|---|
| I:2 | F | 86 | No | 476 | + | – |
| II:1 | M | 67 | No | 446 | + | – |
| II:3 | M | 65 | No | 461 | + | – |
| II:4 | F | 64 | No | 474 | + | – |
| II:6 | F | 62 | No | 467 | + | – |
| II:9 | M | 57 | No | 483 | + | – |
| III:2 | F | 28 | No | 446 | + | – |
| II:7 | M | 63 | No | 432 | – | + |
| III:5 | M | 10 | Sí | 440 | + | + |
| III:6 | F | 39 | No | 416 | – | + |
| IV:1 | M | 5 | No | 406 | – | + |
F, female; M, male; RyR2, ryanodine receptor gene.
Beta-blockers were prescribed to carriers of either mutation, and drugs that prolong QT interval were contraindicated for carriers of the KCNQ1 mutation. At the time of writing, all carriers have remained asymptomatic except for the proband, who is the only carrier of both mutations and had an appropriate implantable cardioverter-defibrillator discharge when aged 19 years.
In summary, the phenotype associated with the p.L3778F RyR2 mutation in this family is probably not as severe as initially suspected. However, until further studies are conducted, it is premature to discount a pathogenic effect for this mutation, especially given that exercise stress test results can be normal in individuals with CPVT.4 The disease severity in the proband, and probably also in his deceased brother, is likely due to the presence of both mutations. The DHPLC-Sanger method can give false negatives, a limitation overcome with genetic screening by NGS.5 The current study highlights the incomplete penetrance and variable expression of LQTS1,3,6 the value of genetic testing for detecting asymptomatic carriers,3,6 and the importance of intercenter cooperation to ensure a complete family study.
FUNDINGThis study was part funded by the Red de Investigación Cardiovascular (RIC) (RD12/0042/0069, RD12/0042/0049).