Noonan syndrome (NS, OMIM 163950) is a genetic multisystem condition with a relatively high estimated incidence of about 1 in 1000 to 1 in 2500 live births.1
This syndrome constitutes the most common syndromic cause of congenital heart disease after Down syndrome.1 The diagnosis of NS depends mainly on the identification of characteristic clinical features, such as a distinctive facial appearance, short stature, and congenital heart disease.1 Cardiovascular abnormalities occur in 50% to 90% of individuals with NS, with pulmonary valve stenosis being the most common. Hypertrophic cardiomyopathy (HCM), found in 20% to 30% of individuals, usually develops early in life. Other cardiovascular abnormalities described in NS include atrial and ventricular septal defects, coarctation of the aorta, partial atrioventricular canal, and tetralogy of Fallot.1
NS has been classically considered an autosomal dominant disorder; however, an autosomal recessive pattern of inheritance related to biallelic variants in the leucine-zipper-like transcriptional regulator 1 (LZTR1) has been very recently described.2 Here we present 2 novel NS cases with autosomal recessive inheritance.
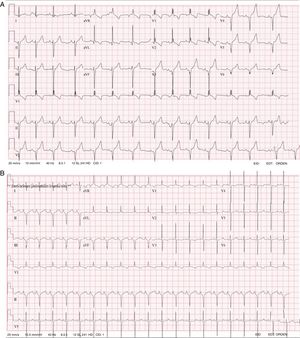
Case 1. A male patient was diagnosed at birth with severe HCM and mild pulmonary valve stenosis. He had the characteristic facies of NS, with broad forehead, hypertelorism, downward-slanting palpebral fissures, posteriorly rotated ears with a thickened helix, and broad thorax with webbed neck. Electrocardiogram (ECG) showed broad QRS complexes for his age (above 0.10seconds at age 4 years), right bundle branch block, left axis deviation, and a striking negative pattern in the left precordial leads (Figure 1A). A genetic test for RASopathies by next-generation sequencing (NGS; 18 genes panel) was requested, which identified 2 novel variants in LTRZ1 (p.Arg362* and c.1149+1G>T).

Case 2. This male patient was diagnosed at birth with severe HCM without obstruction. The ECG was typical of NS, with broad QRS complexes for his age, left axis deviation, and a negative pattern in the precordial leads (Figure 1B). He presented with severe feeding problems, needing a nasogastric tube and a gastrostomy. Genetic testing with the same RASopathies panel was performed and identified 2 novel variants in LTRZ1 (p.Val579Met and c.2070-2A>G).
Genotyping of healthy nonconsanguineous parents confirmed genetic segregation in both clinical cases with biallelic variants.
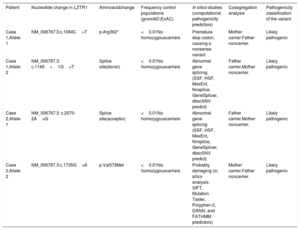
Clinical and genetic analyses allowed classification of the 4 variants found in LZTR1 in the 2 probands presented here as likely pathogenic (Table 1). One of the variant identified in case 1, c.1149+1G>T, has not been previously described in the literature, but the same splice site has been described as being affected in compound heterozygosity in a patient with an AR form of NS2. The other mutation identified in case 1, c.1084C>T (p.Arg362*), introduces a stop signal that leads to an aberrant transcript and would therefore not be translated. As for case 2, c.2070-2A>G affects the canonical splice site in the intronic region of the gene. Finally, the other variant identified in case 2, p.Val579Met, is located in the BACK I domain, where other pathogenic missense mutations have been identified. Future functional studies are needed to definitely confirm their pathogenicity.
Classification of variants identified in LZTR1
| Patient | Nucleotide change in LZTR1 | Aminoacidchange | Frequency control populations (gnomAD;ExAC) | In silico studies (computational pathogenicity predictors) | Cosegregation analysis | Pathogenicity classification of the variant |
|---|---|---|---|---|---|---|
| Case 1,Allele 1 | NM_006767.3:c.1084C>T | p.Arg362* | <0.01No homozygouscarriers | Premature stop codon, causing a nonsense variant | Mother carrier.Father noncarrier. | Likely pathogenic |
| Case 1,Allele 2 | NM_006767.3: c.1149+1G>T | Splice site(donor) | <0.01No homozygouscarriers | Abnormal gene splicing (SSF, HSF, MaxEnt, Nnsplice, GeneSplicer, dbscSNV predict) | Father carrier.Mother noncarrier. | Likely pathogenic |
| Case 2,Allele 1 | NM_006767.3: c.2070-2A>G | Splice site(acceptor) | <0.01No homozygouscarriers | Abnormal gene splicing (SSF, HSF, MaxEnt, Nnsplice, GeneSplicer, dbscSNV predict) | Father carrier.Mother noncarrier. | Likely pathogenic |
| Case 2,Allele 2 | NM_006767.3:c.1735G>A | p.Val579Met | <0.01No homozygouscarriers | Probably damaging (in silico analysis: SIFT, Mutation Taster, Polyphen-2, DANN, and FATHMM predictors) | Mother carrier.Father noncarrier. | Likely pathogenic |
The 2 cases presented here are of special interest for clinical diagnosis and genetic counselling.
First of all, we report 2 novel clinical cases of an autosomal recessive form of NS. This pattern of inheritance was suggested 5 decades ago by Dieckman et al.,3 who described 2 brothers and a sister with clinical features of NS consisting of HCM and pterygium colli, with both parents being unaffected. However, it was not until very recently that clinical and genetic data confirmed the existence of a form of NS inherited following an autosomal recessive pattern, when Johnston et al.2 described biallelic pathogenic variants in LZTR1 in 23 children with clinical NS and with heterozygous, clinically-unaffected parents. LTZR1 germline mutations associated with autosomal dominant NS with a highly variable expressivity had been previously described.4 These data suggest that LZTR1 germline variants could be cause dominant or recessive NS.
Another aspect worth highlighting about the reported cases is that the 2 patients presented with ECG features that had been described decades ago as typical of NS, characterized by wide QRS complexes, with a predominantly negative pattern in the left precordial leads, left axis deviation, and Q waves.5 Other authors found this ECG pattern in almost 60% of NS patients regardless of the presence or absence of anatomical cardiac defects.6 This suggests that ECG is still a very useful tool to support the diagnostic suspicion of Noonan syndrome and should be performed in all patients with NS phenotype.
It is worth looking for any clues that could corroborate suspicion of this syndrome, as NS remains a clinical diagnosis based on the observation of key features, considering that as many as 25% of tested patients lack a genetic molecular basis. However, many of these cases can be expected to have genetic confirmation in the near future by extending molecular testing to these newly described pathogenic variants.
CONFLICTS OF INTERESTL. Monserrat is a stakeholder in Health in Code S.L. J.P. Trujillo-Quintero works at Health in Code SL. The other authors have indicated they have no potential conflicts of interest to disclose.