Marfan syndrome is mainly caused by mutations in the FBN1 gene. Diagnosis is usually based on clinical criteria, but the phenotypic presentation varies widely among affected individuals. Aortic dissection or rupture is the cause of death in over 90% of untreated patients. Early identification of individuals at risk is important given the availability of medical and surgical treatment that can significantly improve life-expectancy. Molecular testing could provide an etiologic diagnosis in patients who present with milder or atypical clinical forms of the disease. Moreover, it could contribute to preventive treatment in carriers, inform genetic counseling, and offer reassurance to unaffected individuals. By describing a family with Marfan syndrome in whom the disease presented in an atypical aggressive form, this article highlights the value of testing for FBN1 mutations in selected cases.
Keywords
Aortic dissection or rupture is the main cause of death in over 90% of untreated patients with Marfan syndrome (MFS),1 but in about 18% the first aortic event (i.e. dilatation or dissection) occurs in the distal aorta.2
In general, MFS is caused by mutations in the fibrillin-1 (FBN1) gene, a large gene located on chromosome 15. More than 600 pathogenic mutations were reported in the last update of the Universal Marfan Database, and most of these are unique to individual families.3
To date, there is no established genotype–phenotype correlation. Nevertheless, the relative risk of specific organ involvement differs significantly between different types of mutations.4
Early recognition of at-risk MFS patients is important in view of available medical and surgical treatments that can significantly improve life-expectancy.
In this paper, we describe a family that includes several members with clinical features compatible with MFS but who have peculiar phenotypic characteristics. In this family, molecular diagnosis provided a definitive diagnosis and could be used for family screening.
Methods Clinical DataPatients and their relatives were invited to a medical consultation with a cardiologist, a rheumatologist and an ophthalmologist; MFS was diagnosed according to Ghent's criteria.5
Echocardiographic evaluations were performed using either a Vivid 3 (GE Healthcare) or iE33 (Philips) ultrasound scanner. The diameter of the aorta was measured on the basis of recommendations for chamber quantification from the American Society of Echocardiography and the European Association of Echocardiography.6 The aortic root was considered dilated when the maximum diameter at the sinuses of Valsalva exceeded the upper normal limit for the patient's age and body surface area.7
Magnetic resonance angiography (MRA) using a 3-T whole-body magnet (Siemens Magnetom Trio) was performed in some patients. Transverse, longitudinal, coronal and oblique sagittal scans encompassing the ascending aorta, aortic arch, and descending aorta were acquired. Multiplanar reconstructions were also analyzed.
The only ocular criterion considered was ectopia lentis. Magnetic resonance was not used to look for dural ectasia.
Molecular DataAfter appropriate written informed consent was obtained, DNA was extracted from peripheral blood samples and all 65 exons of FBN1 were amplified. Mutation screening of the entire coding sequence of the FBN1 gene was then performed using the polymerase chain reaction. All positive results were confirmed by repeated analysis.
Results Family DescriptionOur proband (Figure 1, III:7, arrowed), a 37-year-old female, was referred to our center after experiencing a type-B aortic dissection that started 60mm after the origin of left subclavian artery and extended distally to the iliac arteries. The dimensions of the ascending aorta and aortic arch were within normal limits, while the descending thoracic aorta had a diameter of 42mm.

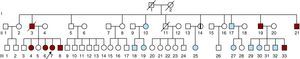
Figure 1. Family pedigree. Arrow, proband. Red-colored symbols, affected subjects fulfilling Ghent criteria. Symbols with a vertical black bar, affected subjects not fulfilling Ghent criteria. Blue symbols, negative clinical and molecular study findings. ?, possibly affected subject. Symbols with an oblique bar, deceased subjects.
Family history at that time revealed that the patient's father (Figure 1, II:3) had died suddenly at the age of 48 years (after a type-B aortic dissection) and two uncles (Figure 1, II:19 and II:21) were already being monitored for aortic disease. The older of these, a 53-year-old male (Figure 1, II:19), had a Bentall composite graft implanted at the age of 50 years because of progressive aortic root dilatation (60mm). The second uncle, who was 48 years old (Figure 1, II:21), presented with generalized arterial disease. His left leg had been amputated because of occlusive arterial disease at the age of 45 years, and a popliteal aneurysm on the right leg had been excluded when he was aged 46 years. He also had mild dilatation of the abdominal aorta and an aortic root aneurysm, of diameters 34mm and 46mm, respectively.
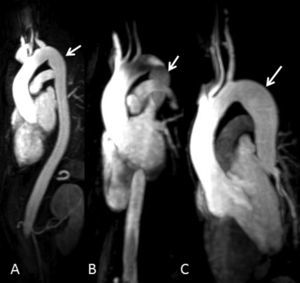
By that time, family screening had been started and our patient's brothers were called in for clinical evaluation. Three of them (two females and one male) were evaluated: a 43-year-old sister (Figure 1, III:4) had a normal-sized aorta but peculiar post-isthmic ectasia was found on MRA (Figure 2A); a 39-year-old sister (Figure 1, III:6) had mild aortic root dilatation (40mm), a normal-sized descending aorta and the same peculiar post-isthmic ectasia (Figure 2B); and the youngest brother, a 36-year-old patient (Figure 1, III:8), presented with aortic root dilatation (47mm) and discrete aortic ectasia (31mm) of the descending aorta just after the isthmus (Figure 2C). Four days after clinical and imaging evaluations, this patient was admitted for acute type-B aortic dissection, which started immediately after the origin of the subclavian artery. A CT scan revealed aortic dilatation (44mm) at the level of the aortic dissection.

Figure 2. Three-dimensional reconstruction of magnetic resonance angiography images of patients III:4 (A), III:6 (B) and III:8 (C) in Figure 1 , showing peculiar mild narrowing at the level of the isthmus and post-isthmic aortic ectasia (arrows).
A first-degree cousin of the patient, a 19-year-old (Figure 1, III:33), had acute type-A dissection at the age of 19 years, which extended throughout the entire aorta. The ascending aorta was markedly dilated (82mm) and he underwent emergent surgical aortic root and valve replacement.
Another of the proband's aunts, a 61-year-old (Figure 1, II:14), was found to have aortic root dilatation (43mm).
The clinical features of affected family members are depicted in Table 1. The other family members evaluated (i.e. 12 subjects) showed no clinical or molecular evidence of MFS.
Table 1. Clinical Features of Affected Family Members.
| Patient | Sex | Age (years) | Cardiovascular system | Skeletal system | Ocular system (ectopia lentis) | Lung (spontaneous pneumothorax) | Skin | ||||||||||
| ADil | ADis | MVP | PCar | PExc | ASp/H | Scol | W&T | PPlan | PAcet | JointH | FacF | ||||||
| II:14 | F | 61 | + | − | − | − | − | 1.03 | − | − | − | na | − | − | na | − | − |
| II:19 | M | 53 | +/Surg | − | +/Surg | na | na | 1.08 | + | + | + | + | − | − | − | − | − |
| II:21 | M | 48 | + | − | − | na | na | 1.03 | − | + | − | + | − | + | − | + | + |
| III:4 | F | 43 | − | − | + | − | + | 1.04 | + | + | + | + | − | + | − | − | + |
| III:6 | F | 39 | + | − | − | − | + | 1.02 | +/Surg | + | + | + | + | − | − | + | + |
| III:7 | F | 37 | − | + | + | − | + | 1.04 | + | + | + | + | + | + | − | − | + |
| III:8 | M | 36 | + | + | + | + | − | 1.06 | + | + | + | + | + | − | − | − | + |
| III:33 | M | 19 | + | +/Surg | − | − | − | 1.08 | − | + | − | + | + | + | − | − | + |
Abbreviations: ADil, aortic dilatation; ADis, aortic dissection; Asp/H, arm-span-to-height ratio; F, female; FacF, facial features; JointH, joint hypermobility; M, male; MVP, mitral valve prolapse; na, not available; PAcet, protusio acetabulae; PCar, pectus carinatum; PExc, pectus excavatum; PPlan, pes planus; Scol, scoliosis; Surg, surgery; W&T, wrist and thumb signs; −, not present; +, present.
A new pathogenic FBN1 mutation was identified in all affected members of this family. The mutation was a nonsense mutation, with a C1995G substitution in exon 16 of FBN1, which led to the formation of a stop codon in position 665 of tyrosine (i.e. Tyr665X).
DiscussionThis family came to our attention because of two cases of type-B aortic dissection (Figure 1, III:7 and III:8). The aortic diameters were smaller than expected in these complications and the descending aorta had a peculiar anatomy, which had not previously been described as being associated with MFS (Figure 2). Moreover, another family member presented with an initial diagnosis of peripheral arterial disease (i.e. a popliteal aneurysm) and was subsequently diagnosed with abdominal and ascending aortic dilatation. The presence of a generalized arterial familial syndrome was suspected and we searched for mutations in FBN1 and transforming growth factor-beta receptor (TGFβR) genes. The molecular identification of a FBN1 mutation was a determinant of a definitive diagnosis of MFS in this family.
In general, FBN1 gene mutations are found in about 90% of MFS patients who fulfill Ghent's criteria,8 but patients with classic MFS phenotypes have also been found to be carriers of TGFβR1 and TGFβR2 mutations (MFS type 2), the same mutations that cause Loeys–Dietz syndrome (LDS). These patients typically present at an early age with generalized arterial tortuosity and aneurysms and aortic dissection and have smaller aortic diameters.9 A broad clinical overlap has been found among MFS, MFS type 2 and LDS.10
In this family, we found a new FBN1 mutation that leads to an early protein sequence truncation and which is associated with a particularly aggressive cardiovascular phenotype in most affected individuals. The nonsense mutation found was located in the exon encoding the second of seven TGFβ1-binding protein-like modules of fibrillin-1, a domain present only in FBN1 and latent TGF-beta binding proteins (LTBPs). These globular domains have no known specific function, although they may mediate specific protein–protein interactions.11
Currently, increased TGFβ activity is thought to be a major contributor to the pathogenesis of both MFS and LDS.9,12
In the future, better understanding of the genetic background and molecular pathways involved may change our approach to aortic disease in MFS. Meanwhile, currently accepted criteria for surgical aortic intervention in MFS patients are an ascending aorta diameter of 5.0cm or more and a descending aorta diameter of at least 6.0cm.13 These values are far from the aortic dimensions presented by our proband and her brother, yet this did not prevent them developing type-B aortic dissection. The questions of whether and when to intervene in the other two sisters who have similar aortic morphologies are difficult, and these individuals are now under close clinical and imaging surveillance to detect any increase in aortic dimensions. In conclusion, in this family, detection of a FBN1 mutation revealed the genetic nature of the phenotypic features described, enabled patients with milder clinical presentations to be diagnosed, and played a role in early diagnosis, genetic counseling, preventive management of carriers and reassurance of unaffected individuals. The differential diagnosis of classic MFS, MFS type 2 and an LDS subtype would not have been possible on clinical grounds alone.
FundingThis study was supported by a grant from the Research Department of Hospital S. João, Porto, Portugal.
Conflicts of interestNone declared.
Received 2 January 2010
Accepted 21 February 2010
Corresponding author: Hospital de S. João, Alameda Prof. Hernâni Monteiro 4202-451 Porto, Portugal. ana.lebreiro@gmail.com