El síndrome de QT largo congénito tiene su causa principal en mutaciones de los genes KCNQ1, KCNH2 y SCN5A. Nos proponemos analizar la prevalencia de mutaciones en estos genes en nuestra serie de pacientes con síndrome de QT largo y fibrilación ventricular idiopática. Se incluyó a 9 pacientes con síndrome de QT largo y 4 con fibrilación ventricular idiopática. Se estudió a los familiares de primer grado de los probandos con genotipo positivo. Encontramos mutaciones missense en 7 pacientes con síndrome de QT largo y en 2 con fibrilación ventricular idiopática. El 71,4% de las mutaciones fueron en KCNH2 y el 28,6% en SCN5A. No se halló ninguna mutación en KCNQ1. Sólo dos mutaciones estaban previamente descritas. En 6 familiares de los 19 estudiados se encontró una mutación. En conclusión, en nuestra experiencia inicial el estudio genético tuvo una alta sensibilidad para el diagnóstico de síndrome de QT largo. El gen más frecuentemente mutado fue KCNH2.
Palabras clave
El síndrome de QT largo (SQTL) es una canalopatía cardiaca que puede causar muerte súbita por arritmias ventriculares. Se han descrito cientos de mutaciones en doce genes de canales de sodio y potasio principalmente1,2. Aproximadamente el 75% de las mutaciones descritas en el SQTL se encuentran en tres genes: KCNQ1 (canal de potasio), KCNH2 (canal de potasio) y SCN5A (canal de sodio)1. Un 25–30% de los pacientes con SQTL permanecen sin diagnóstico genético a pesar de la secuenciación completa de todos los genes descritos3.
El SQTL tiene una penetrancia de un 25–90%, por lo que existe la posibilidad de padecer la enfermedad con un electrocardiograma normal. En caso de parada cardiaca, estos pacientes pueden catalogarse como fibrilación ventricular idiopática (FVI). Aunque la etiología de este síndrome no sólo implica a las canalopatías, recientemente se ha señalado la posible utilidad del estudio de los genes implicados en el SQTL4.
El objetivo principal de este trabajo es describir las características básicas del genotipo en nuestra serie de pacientes con SQTL y la utilidad de este análisis genético en los casos con FVI.
MétodosSe incluyó a 9 pacientes con criterios diagnósticos de SQTL (media de edad, 22,6±21,6 años; el 66,7% mujeres) y 4 con FVI (26±22,1 años; el 50% mujeres) estudiados en nuestra unidad de arritmias. El estudio incluyó historia familiar, analítica, ecocardiograma y Holter. En los 4 pacientes con FVI e intervalo QT corregido (QTc) normal, además se realizó estudio electrofisiológico, coronariografía y test de provocación con flecainida y adrenalina. Realizamos estudio genético en 19 familiares de los probandos con genotipo anormal. Todos los pacientes o tutores firmaron un consentimiento informado.
Mediante la técnica de reacción en cadena de la polimerasa, se secuenciaron los genes en una muestra de 5 ml de sangre periférica. Para considerar patológico un hallazgo, se exigió una mutación missense y que dicha alteración no estuviese presente en el grupo control.
ResultadosLas características clínicas de los pacientes se muestran en la Tabla 1. Los resultados del estudio clínico y genético se muestran en la Tabla 2. De los 9 pacientes con SQTL, 7 (77,7%) presentaban mutaciones: el 71,4% de éstas en KCNH2 y el 28,6% en SCN5A. No encontramos ninguna mutación en KCNQ1. Únicamente dos mutaciones estaban ya descritas como causa de SQTL2,5. Dos enfermos (50%) con FVI presentaron mutaciones, uno en KCNH2 y otro en SCN5A, que tampoco estaban previamente descritas.
Tabla 1. Características clínicas de los pacientes de QT largo y fibrilación ventricular idiopática incluidos en el estudio
| Sexo | Edad al primer síntoma | Historia de síncope | Edad (años) al diagnóstico | Síntoma que motiva el diagnóstico | Diagnóstico fenotípico | PCR o descarga DAI | Desencadenantes | |
| Caso 1 | M | 11 | Sí | 49 | PCR | SQTL | Sí | Ruidos |
| Caso 2 | M | 60 | No | 67 | PCR | SQTL | Sí | Emociones |
| Caso 3 | M | 14 | Sí | 23 | PCR | SQTL | Sí | Emociones |
| Caso 4 | V | 17 | No | 24 | PCR | FVI | Sí | Natación |
| Caso 5 | M | 10 | Sí | 11 | PCR | FVI | Sí | Despertar |
| Caso 6 | V | 9 | Sí | 11 | Síncope | SQTL | No | Emociones |
| Caso 7 | M | 53 | Sí | 58 | PCR | FVI | Sí | Desconocido |
| Caso 8 | M | 0 | No | 1 día | PCR | SQTL | Sí | Descanso |
| Caso 9 | M | 19 | Sí | 20 | Síncope | SQTL | No | Descanso |
| Caso 10 | V | 2 | Sí | 10 | Síncope | SQTL | No | Desconocido |
| Caso 11 | M | 12 | Sí | 18 | PCR | SQTL | Sí | Emociones |
| Caso 12 | V | 10 | Sí | 11 | Síncope | FVI | Sí | Desconocido |
| Caso 13 | V | No | No | 5 | Asintomático | SQTL | No | No |
DAI: desfibrilador automático implantable; FVI: fibrilación ventricular idiopática; M: mujer; PCR: parada cardiorrespiratoria; SQTL: síndrome de QT largo; V: varón.
Tabla 2. Espectro mutacional del síndrome de QT largo en nuestra serie de pacientes
| QTc | Test farmacológico (epinefrina/flecainida) | Diagnóstico fenotípico | Score diagnóstico | Tratamiento | Estudio genético | Diagnóstico final | |
| Caso 1 | 555 | No | SQTL | 8 | DAI | KCNH2+(1910 A>G Glu>Gly) | SQTL2 |
| Caso 2 | 478 | No | SQTL | 4 | DAI | SCN5A+(3985 G>R Gly>Ser) | SQTL3 |
| Caso 3 | 442 | + | SQTL | 4 | DAI | SCN5A+(1673 A>R His>Arg) | SQTL |
| Caso 4 | 367 | − | FVI | 1 | DAI | Negativo | FVI |
| Caso 5 | 405 | − | FVI | 2 | DAI | KCNH2+(541 C>Y Arg>Tpf) (577 G>R Al>Thr) (62 G>R Gly>Asp) | SQTL2 |
| Caso 6 | 631 | No | SQTL | 8 | DAI | KCNH2+(1714 G>R Gly>Ser) | SQTL2 |
| Caso 7 | 413 | − | FVI | 2 | DAI | SCN5A+(1673 A>R His>Arg) | FVI |
| Caso 8 | 720 | No | SQTL | 7,5 | DAI | Negativo | SQTL |
| Caso 9 | 577 | No | SQTL | 4 | BB | Negativo | SQTL |
| Caso 10 | — | No | SQTL | 4 | DAI | KCNH2+(343G>R Val>Met) | SQTL2 |
| Caso 11 | 488 | + | SQTL | 5 | DAI | KCNH2+(1882 G>S Gly>Arg) | SQTL2 |
| Caso 12 | 400 | − | FVI | 1 | DAI | Negativo | FVI |
| Caso 13 | 476 | No | SQTL | 3,5 | No | KCNH2+(del 3079-3124) | SQTL2 |
A: adenina en homocigosis; Al: alanina; Arg: arginina; Asp: ácido aspártico; BB: bloqueadores beta; C: citosina en homocigosis; DAI: desfibrilador automático implantable; FVI: fibrilación ventricular idiopática; G: guanina en homocigosis; Gly: glicina; Glu: ácido glutámico; His: histidina; Met: metionina; R: guanina/adenina en heterocigosis; S: guanina/citosina en heterocigosis; Ser: serina; SQTL: síndrome de QT largo; Thr: treonina; Tpf: triptófano; Val: valina.
Este es el gen con más frecuencia de mutaciones. La media de edad fue 17,3±16 años y el intervalo QTc medio, 511 ms. Encontramos ocho mutaciones (Tabla 2), sólo dos de ellas descritas previamente como causa de SQTL (G1882S y G1714R)2,5. Ambas están localizadas entre la región P (poro) y el dominio transmembrana 5, una región fundamental como filtro de selectividad de potasio6.
Gen SCN5AEl 28,6% de los casos de SQTL genéticamente positivos presentó mutaciones missense no descritas previamente en este gen. Una paciente con FVI presentó una mutación para la que no hay evidencia de implicación causal directa en el SQTL. Sin embargo, en la evolución clínica hay datos que respaldan el diagnóstico de SQTL tipo 3; por un lado, ha tenido descargas del desfibrilador automático implantable, con registro de episodios de torsade de pointes, y por otro, ha presentado episodios de fibrilación auricular paroxística con respuesta a flecainida.
Valor diagnóstico y pronóstico del estudio genético en nuestro medio. Utilidad en la fibrilación ventricular idiopáticaBasalmente había 5 pacientes sin cardiopatía e intervalo QTc normal. El test farmacológico con adrenalina fue normal en 4 de ellos y diagnóstico de SQTL en 1 caso que, además, luego tuvo test genético positivo. En 2 de los 4 pacientes con FVI e intervalo QTc basal y tras fármacos dentro de la normalidad, el estudio genético mostró mutaciones missense.
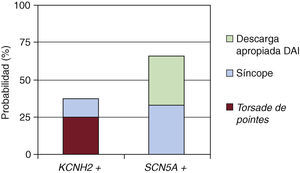
El registro de eventos sufridos por los enfermos con bloqueadores beta y en función del canal afecto se muestra en la Figura 1. Tres enfermos con mutación en KCNH2 presentaron episodios: 2 casos taquicardia ventricular polimórfica tipo torsade de pointes y 1 síncope, frente a 2 con mutación en SCN5A: 1 síncope y 1 descarga apropiada de desfibrilador automático implantable.

Figura 1. Probabilidad de sufrir un evento (síncope, descarga apropiada del desfibrilador automático implantable [DAI] o torsade de pointes) tras tratamiento con bloqueador beta (p>0,05).
Estudio genético en familiares asintomáticosSe ha estudiado a 19 familiares directos de los enfermos con mutaciones, y se encontró la mutación del caso índice en 6 (31,5%), con fenotipo negativo en 2 de ellos (portadores silentes).
En los 2 casos con mutaciones ya descritas, el estudio genético fue negativo en los 7 familiares estudiados; todos ellos tenían un QTc normal, lo que nos ha permitido descartar la enfermedad.
DiscusiónNo hay datos en nuestro medio acerca de las características genéticas del SQTL en series de pacientes, y se han publicado únicamente mutaciones aisladas7. Nuestro trabajo es una experiencia preliminar e inicial en nuestro país, y describe el perfil genotípico de nuestra pequeña muestra de pacientes con SQTL y FVI. Es necesaria una colaboración multicéntrica para obtener grupos más amplios y conclusiones extrapolables a la población general. Estudios previos demuestran que en un 65–70% de los casos se detecta alteración genética, y el gen más frecuentemente mutado es KCNQ11,8. Nosotros hemos encontrado una frecuencia de mutaciones algo superior (77,7%), y en nuestra serie KCNH2 es el gen más frecuentemente afectado. Estos resultados no se pueden generalizar debido al pequeño tamaño muestral.
De las nueve mutaciones missense encontradas, únicamente dos estaban previamente descritas como causa de SQTL. En los familiares asintomáticos de dichos pacientes fue posible descartar la enfermedad, pero para establecer de manera incontestable la patogenicidad del resto de las mutaciones, sería preciso un estudio de las propiedades biofísicas del canal iónico afectado con esa mutación. Sin embargo, ciertos datos, como la zona del canal afecto, el cambio aminoacídico resultante y la ausencia de esta proteína en los controles, orientan sobre la patogenicidad de la mutación9.
En nuestra serie, el 22,2% de los enfermos con SQTL y el 50% de los casos de FVI permanecieron con genotipo negativo. Ello subraya la necesidad de extender el análisis genético en los casos negativos al resto de los genes descritos, así como la importancia de explorar nuevos genes candidatos para explicar la causa del SQTL10,11.
La proporción de eventos arrítmicos tras tratamiento con bloqueadores beta, mayor en los SQTL3, es elevada; la razón fundamental es el sesgo de selección por tratarse de población en alto riesgo reclutada en ámbito hospitalario, con 8 de los 13 casos reanimados tras una parada cardiaca.
Dos de los 4 pacientes con FVI presentaron mutaciones. Aunque no podemos saber el impacto patogénico de estas mutaciones, uno de los casos presentó tres mutaciones en la región N-terminal de KCNH2, con alta probabilidad de alterar la corriente de potasio a través del canal. Nuestro trabajo sugiere que el test genético tiene utilidad diagnóstica en los pacientes supervivientes a una fibrilación ventricular en los que no se llegue al diagnóstico tras un protocolo de estudio completo, incluso tras test farmacológicos negativos.
El 31,5% de los familiares estudiados eran portadores de la mutación y el 33% de éstos, portadores silentes. Aunque todavía es incompleto el conocimiento acerca del pronóstico de los portadores silentes, es obligado prevenirlos sobre el uso de fármacos que prolonguen el intervalo QT y valorar la iniciación de tratamiento con bloqueadores beta.
En conclusión, en nuestra experiencia inicial, el estudio genético tuvo una alta sensibilidad en nuestra serie de pacientes con SQTL y su resultado difiere del previamente descrito porque el subtipo 2 es el más prevalente. Nuestro trabajo sugiere que el análisis genético de estos tres genes puede ser una prueba útil en la FVI. Estas hipótesis deben ser contrastadas en estudios con mayor tamaño muestral.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Recibido 22 Enero 2010
Aceptado 25 Febrero 2010
Autor para correspondencia. Servicio de Cardiología, Hospital Universitario Virgen de las Nieves, Avda. de las Fuerzas Armadas, 2. 18014 Granada, España. jimenez.jaimez@gmail.com