ISSN: 0300-8932
Factor de impacto 2023
7,2
SEC 2022 - El Congreso de la Salud Cardiovascular

Palma de Mallorca y online,
20 - 22 de Octubre de 2022
Introducción
Dr. Juan José Gómez Doblas
Presidente del Comité Científico del Congreso
Comité ejecutivo
Comité de evaluadores
Listado de sesiones
Índice de autores
5016. Cardiología traslacional: del laboratorio al paciente
Fecha
: 22-10-2022 09:30:00
Tipo
: Comunicaciones mini orales
Sala
: Sala Menorca 2 (Planta 3)
5016-2. EL PAPEL PROTECTOR SOBRE EL ESTRÉS DEL RETÍCULO ENDOPLÁSMICO EN EL CARDIOMIOCITO A TRAVÉS DE LA INHIBICIÓN DEL MIR-16-5P
Elena Alonso Villa1, Fernando Bonet Martínez1, Aníbal Bermúdez2, Tomás Daroca2, Maribel Quezada-Feijoo3, Mónica Ramos3, Alipio Mangas Rojas2 y Rocío Toro Cebada1
1Universidad de Cádiz, 2Hospital Universitario Puerta del Mar, Cádiz y 3Hospital Central de la Cruz Roja, Madrid.
1Universidad de Cádiz, 2Hospital Universitario Puerta del Mar, Cádiz y 3Hospital Central de la Cruz Roja, Madrid.
Introducción y objetivos: El estrés del retículo endoplásmico (ERE) influye en la patogénesis de la cardiomiopatía dilatada (CMD). Este ERE desencadena la activación de la respuesta a proteínas desplegadas (UPR) para restablecer la proteostasis celular, evitando la apoptosis. Sin embargo, cuando el ERE es severo o crónico conduce a la muerte celular. Los microRNA están vinculados en la fisiopatología de varias enfermedades cardiovasculares. Nuestro grupo ha publicado con anterioridad que miR-16-5p está enriquecido en el plasma de pacientes con CMD isquémica (CMI) y que este miR-16-5p induce ERE y daño celular asociado en cardiomiocitos humanos. Nuestro objetivo es investigar si la inhibición de miR-16-5p puede ejercer un papel cardioprotector contra el daño celular asociado al ERE.
Métodos: Cardiomiocitos humanos (AC16) se trataron con tunicamicina (TN), un inductor del ERE, en presencia del inhibidor de miR-16-5p. Posteriormente determinamos la presencia de marcadores de ERE, apoptosis, inflamación y daño cardiaco mediante qPCR y/o cartometría de flujo. Por último, analizamos la regulación de la transcripción de miR-16-5p sobre su gen diana ATF6 mediante ensayo de luciferasa.
Resultados: La inhibición de miR-16-5p revertió el daño celular asociado al ERE inducido por la TN en células AC16. La inhibición de miR-16-5p revertió la regulación negativa de ATF6 inducida por la TN (fig. 1A). Además, la inhibición de miR-16-5p rescató los niveles de expresión de los genes antioxidantes CAT y SOD1 (fig. 1B), del gen antiapoptótico BCL-2 (fig. 1C), y del gen regulador de la autofagia ATG14 (fig. 1D). Además, la inhibición de miR-16-5p recupera los niveles de expresión de IL-6 y TNNT2, marcadores de inflamación y de daño del cardiomiocito, respectivamente (fig. 1E). Por último, ensayos de luciferasa demuestran que miR-16-5p se une al sitio diana predicho del extremo 3’UTR de ATF6 (fig. 1F).
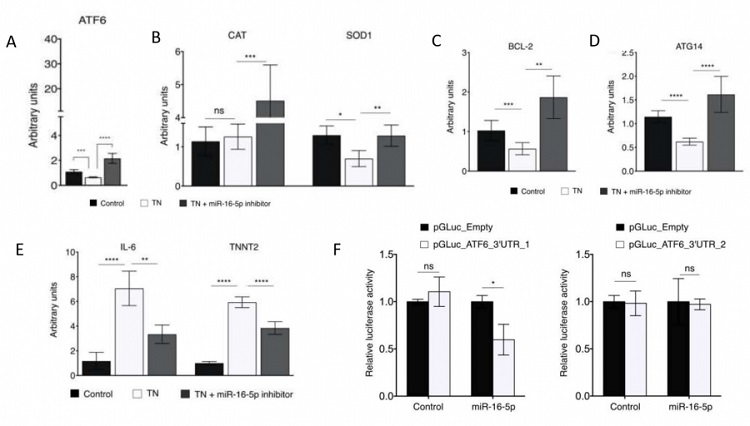
La inhibición de miR-16-5p revierte el daño causado por el ERE inducido por TN en cardiomioblastos.
Conclusiones: Nuestros resultados confirman que miR-16-5p juega un papel crítico en la regulación del ERE modulando la expresión de ATF6 en cardiomiocitos humanos, y que su inhibición protege a los cardiomiocitos humanos del daño celular asociado al ERE induciendo la vía citoprotectora mediada por ATF6. Por tanto, la supresión de miR-16-5p puede ser una estrategia terapéutica contra el desarrollo y progresión de la CMI.
Comunicaciones disponibles de "Cardiología traslacional: del laboratorio al paciente"
- 5016-1. MODERADOR
- Francisco Javier Roselló Lozano, Palma
- 5016-2. EL PAPEL PROTECTOR SOBRE EL ESTRÉS DEL RETÍCULO ENDOPLÁSMICO EN EL CARDIOMIOCITO A TRAVÉS DE LA INHIBICIÓN DEL MIR-16-5P
- Elena Alonso Villa1, Fernando Bonet Martínez1, Aníbal Bermúdez2, Tomás Daroca2, Maribel Quezada-Feijoo3, Mónica Ramos3, Alipio Mangas Rojas2 y Rocío Toro Cebada1
1Universidad de Cádiz, 2Hospital Universitario Puerta del Mar, Cádiz y 3Hospital Central de la Cruz Roja, Madrid.
- 5016-3. INFLUENCIA DE MIR-16-5P EN EL ESTRÉS OXIDATIVO Y LA DISFUNCIÓN MITOCONDRIAL EN CARDIOMIOCITOS
- Elena Alonso Villa1, Fernando Bonet Martínez1, Aníbal González2, Tomás Daroca2, Maribel Quezada-Feijoo3, Mónica Ramos3, Alipio Mangas Rojas2 y Rocío Toro Cebada1
1Universidad de Cádiz, 2Hospital Universitario Puerta del Mar, Cádiz y 3Hospital Central de la Cruz Roja, Madrid.
- 5016-4. PROTECCIÓN CARDIACA DE LA PIRFENIDONA POSINFARTO: UN ANÁLISIS BIOINFORMÁTICO
- Oriol Iborra Egea1, Alberto Aimo2, Nicola Martini3, Carolina Gálvez Montón1, Silvia Burchielli4, Giorgia Panichella2, Claudio Passino4, Michele Emdin2 y Antoni Bayés Genís5
1Fundació Institut en Ciències de la Salut Germans Trias i Pujol, Badalona, Barcelona, 2Fondazione Toscana Gabriele Monasterio, Pisa, Toscana, 3Scuola Superiore Sant’Anna, Pisa, Toscana, 4Scuola Superiore Sant’Anna, Pisa, Toscana y 5Hospital Universitari Germans Trias i Pujol, Badalona (Barcelona).
- 5016-5. CORRELACIÓN ENTRE LIPOPROTEÍNA A Y CARGA ATEROSCLERÓTICA MEDIANTE SOFTWARE DE RECONSTRUCCIÓN CORONARIA TRIDIMENSIONAL
- Pablo Manuel Fernández Corredoira, Carlos Cortes-Villar, Victoria Marco Benedí, Esther Sanz Patiño, Elisa García Arceiz, Darío Javier Samaniego Pesantez, Marta Antonio Martín, Araceli Sánchez Page, David Gómez Martín, Isabel Ezpeleta Sobrevía, Luis Cerdán Ferreira, Teresa Simón Paracuellos, Vanesa Alonso Ventura, Gabriel Hurtado Rodríguez y M. del Rosario Ortas Nadal
Hospital Universitario Miguel Servet, Zaragoza.
- 5016-6. ANÁLISIS DE LA EXPRESIÓN DE LA PROTEÍNA MITOCONDRIAL TRANSPORTADORA DE PIRUVATO EN TEJIDO CARDIACO, DE ORIGEN ISQUÉMICO O NO ISQUÉMICO, DE PACIENTES CON INSUFICIENCIA CARDIACA
- Paula López Vázquez, María G. Crespo Leiro, Eduardo Barge Caballero, Noelia Blanco Menéndez, María Jesús Paniagua Martín, Gonzalo Barge Caballero, David Couto Mallón, Fernanda Regueira Bulgheroni, Zulaika Grillé Cancela, Paula Blanco Canosa, José Manuel Vázquez Rodríguez y Nieves Doménech García
Complexo Hospitalario Universitario A Coruña.
- 5016-7. ANÁLISIS DE LA ORIENTACIÓN DE LAS FIBRAS DE COLÁGENO EN CICATRIZ DE INFARTO AGUDO DE MIOCARDIO MEDIANTE TRANSFORMADA DE FOURIER
- Víctor Marcos Garcés1, César Ríos Navarro2, Fabián A. Gómez Torres3, José Gavara Doñate2, Elena De Dios Lluch4, Nerea Pérez Solé2, Ana Díaz Cuevas5, Gema Miñana Escrivà1, Francisco Javier Chorro Gascó1, Vicente Bodí Peris1 y Amparo Ruiz Sauri6
1Hospital Clínico Universitario de Valencia, 2Fundación de Investigación del Hospital Clínico de Valencia-INCLIVA, 3Universidad Industrial de Santander, Bucaramanga, 4Centro de Investigación Biomédica en Red de Enfermedades Cardiovasculares (CIBER-CV), Madrid, 5Unidad Central de Investigación en Medicina, Universitat de València y 6Departament de Patologia, Universitat de València, Valencia.
- 5016-8. EFECTO INMUNOMODULADOR DEL BIOIMPLANTE PERICORD: DATOS PRELIMINARES DEL ENSAYO CLÍNICO FIRST-IN-HUMAN PERISCOPE
- Paloma Gastelurrutia Soto1, Marta Monguio-Tortajada1, Cristina Prat-Vidal2, María Luisa Cámara-Rosell3, Germán Cediel Calderón3, Elena Revuelta-López3, Albert Téis3, Elena Barceló Cormano3, Santiago Roura1, Carolina Gálvez Montón1, Josep Lupón Rosés3, Anna Vilarrodona2, Christian Muñoz Guijosa3, Luciano Rodríguez Gómez2 y Antoni Bayés Genís3
1Institut Germans Trias i Pujol, Badalona (Barcelona), 2Banc de Sang i Teixits (BST), Barcelona y 3Hospital Universitari Germans Trias i Pujol, Badalona (Barcelona).
- 5016-9. INTERVENCIÓN METABÓLICA PARA EL TRATAMIENTO DEL MIOCARDIO HIBERNADO: ENSAYO PRECLÍNICO FAT4HEART
- Juan Martínez Milla1, Carlos Galán Arriola1, Daniel Pérez Camargo1, Ana Devesa Arbiol1, Rodrigo Fernández Jiménez1, Rocío Villena1, Beatriz Salinas1, Manuel Carnero Alcázar2, Luis Nieto Roca3, Gonzalo Javier López1, Eduardo Oliver1, Valentín Fuster de Carulla1, Manuel Desco4, Javier Sánchez González1 y Borja Ibáñez1
1Centro Nacional de Investigaciones Cardiovasculares (CNIC), Madrid, 2Hospital Clínico San Carlos, Madrid, 3Fundación Jiménez Díaz, Madrid y 4Hospital Gregorio Marañón, Instituto de Investigación Sanitaria Gregorio Marañón, CIBERCV, Madrid.
Más comunicaciones de los autores
- Alonso Villa, Elena
- Bermúdez, Aníbal
- Bonet Martínez, Fernando
- Daroca, Tomás
- Mangas Rojas, Alipio
-
Quezada-Feijoo, Maribel
- 5016-3 - INFLUENCIA DE MIR-16-5P EN EL ESTRÉS OXIDATIVO Y LA DISFUNCIÓN MITOCONDRIAL EN CARDIOMIOCITOS
- 5016-2 - EL PAPEL PROTECTOR SOBRE EL ESTRÉS DEL RETÍCULO ENDOPLÁSMICO EN EL CARDIOMIOCITO A TRAVÉS DE LA INHIBICIÓN DEL MIR-16-5P
- 6041-8 - UTILIDAD DE LOS BIOMARCADORES NTPROBNP Y GALECTINA-3 EN EL MANEJO DE LA ESTENOSIS AÓRTICA GRAVE ASINTOMÁTICA. ESTUDIO DE CASOS Y CONTROLES
-
Ramos Sánchez, Mónica
- 5016-3 - INFLUENCIA DE MIR-16-5P EN EL ESTRÉS OXIDATIVO Y LA DISFUNCIÓN MITOCONDRIAL EN CARDIOMIOCITOS
- 5016-2 - EL PAPEL PROTECTOR SOBRE EL ESTRÉS DEL RETÍCULO ENDOPLÁSMICO EN EL CARDIOMIOCITO A TRAVÉS DE LA INHIBICIÓN DEL MIR-16-5P
- 6041-8 - UTILIDAD DE LOS BIOMARCADORES NTPROBNP Y GALECTINA-3 EN EL MANEJO DE LA ESTENOSIS AÓRTICA GRAVE ASINTOMÁTICA. ESTUDIO DE CASOS Y CONTROLES
-
Toro Cebada, Rocío
- 5016-2 - EL PAPEL PROTECTOR SOBRE EL ESTRÉS DEL RETÍCULO ENDOPLÁSMICO EN EL CARDIOMIOCITO A TRAVÉS DE LA INHIBICIÓN DEL MIR-16-5P
- 5016-3 - INFLUENCIA DE MIR-16-5P EN EL ESTRÉS OXIDATIVO Y LA DISFUNCIÓN MITOCONDRIAL EN CARDIOMIOCITOS
- 6041-8 - UTILIDAD DE LOS BIOMARCADORES NTPROBNP Y GALECTINA-3 EN EL MANEJO DE LA ESTENOSIS AÓRTICA GRAVE ASINTOMÁTICA. ESTUDIO DE CASOS Y CONTROLES