Normal endocrine function is essential for cardiovascular health. Disorders of the endocrine system, consisting of hormone hyperfunction and hypofunction, have multiple effects on the cardiovascular system. In this review, we discuss the epidemiology, diagnosis, and management of disorders of the pituitary, thyroid, parathyroid, and adrenal glands, with respect to the impact of endocrine dysfunction on the cardiovascular system. We also review the cardiovascular benefits of restoring normal endocrine function.
Keywords
Normal endocrine function is essential for cardiovascular health. Disorders of the endocrine system, consisting of hormone hyperfunction and hypofunction, have multiple effects on the cardiovascular system. The objective of this review is to explore the various cardiovascular changes that occur in endocrine dysfunction. We will also assess the cardiovascular benefits of correcting endocrine disorders. Diabetes is specifically excluded, as the well-known relationship between diabetes and cardiovascular risk is beyond the scope of this review.
The pituitary gland and the cardiovascular system Pituitary OverviewThe anterior pituitary gland contains five cell types that synthesize and secrete hormones (growth hormone [GH], prolactin, follicle stimulating hormone, luteinizing hormone, thyroid stimulating hormone [TSH], adrenocorticotropic hormone [ACTH]), that participate in hypothalamic-pituitary-target organ regulation. The posterior pituitary contains nerve terminals that secrete vasopressin (antidiuretic hormone) and oxytocin. Of the pituitary hormones secreted by the anterior pituitary, disorders of prolactin, GH, and ACTH may be associated with cardiac disease.
Prolactin Disorders and Cardiovascular DiseaseProlactin is synthesized and secreted by lactotroph cells of the anterior pituitary gland, and stimulates lactation in the postpartum period. Prolactin is tonically inhibited by hypothalamic dopamine. Prolactin levels are physiologically elevated in pregnancy, the postpartum period, and in states of stress. Pathologic hyperprolactinemia may be caused by decreased dopaminergic inhibition, such as when the pituitary stalk is disrupted, or by prolactin secretion from prolactinomas (benign pituitary adenomas). The prevalence of hyperprolactinemia ranges from 0.4% in the general adult population to 9% in women with reproductive disorders.1 Although hyperprolactinemia itself does not have clear effects on the cardiovascular system, there is a possible association between long-term treatment with dopamine agonists and cardiac valve abnormalities.
Dopamine agonists, including cabergoline, bromocriptime, and quinagolide (not approved for use in the United States), are the primary treatment for prolactinomas. Cabergoline is most commonly used, due to its clinical efficacy, tolerability, and favorable pharmacokinetic profile.2 High doses and long duration of therapy with dopamine agonists have been associated in Parkinson's disease with an increased risk of regurgitant valve disease.3,4 Although doses used for prolactinoma therapy are much lower than those used for Parkinson's disease, patients with prolactinoma may be treated for decades. This treatment duration raises concern for increased risks of valvulopathy, including tricuspid regurgitation, mitral regurgitation, and aortic regurgitation.5,6 Although most reports do not show an association between the use of dopamine agonists and cardiac valve disease, clinicians are advised to use the lowest possible doses of dopamine agonists. Echocardiographic monitoring should be considered, especially in patients requiring long-term and/or higher-dose therapy, and those with underlying heart or valvular disease.7
Peripartum cardiomyopathy is a rare clinical entity. It has been suggested that a 16 kDa prolactin fragment may play a role in its pathophysiology.8 Case reports have described the use of bromocriptine in addition to standard heart failure therapy in peripartum cardiomyopathy.9
Growth Hormone OverviewGH is synthesized and secreted by somatotroph cells in the anterior pituitary gland. It acts directly on peripheral tissues via interaction with the GH receptor, and indirectly via stimulation of insulin-like growth factor type 1 (IGF-1) synthesis. In virtually all cell types, IGF-1 promotes glucose uptake and cellular protein synthesis. GH and IGF-1 regulate somatic growth, including cardiac development and function10
The prevalence of GH deficiency (GHD) in adults is approximately 1-2 per 10,000.11 The prevalence of acromegaly, or excess GH secretion, is approximately 40-70 cases per million, with an estimated incidence of 3-4 per million annually.12,13
Growth Hormone Deficiency OverviewAdults with GHD can be grouped into three categories: those with childhood onset GHD, those with acquired GHD secondary to structural lesions or trauma, and those with idiopathic adult onset GHD.14 Diagnosis is confirmed by low serum IGF-1 levels and provocative testing using insulin-induced hypoglycemia, and the combination of arginine and GH-releasing hormone (GHRH), which are potent stimuli for GH secretion. A subnormal increase in serum GH concentration after insulin tolerance or GHRH-arginine tests confirms the diagnosis of GHD.15 Treatment of GHD consists of GH replacement.
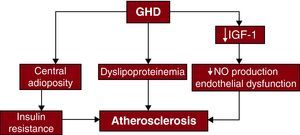
Growth Hormone Deficiency and Cardiovascular Disease Cardiovascular RiskGHD is associated with increased body fat and central adiposity, dyslipidemia (low high density lipoprotein cholesterol [HDLc], high total cholesterol, and high low density lipoprotein cholesterol [LDLc]), endothelial dysfunction, and insulin resistance16,17 (Figure 1). Increased carotid arterial intima-media thickness (IMT), a marker of early atherosclerotic development, has also been described in GHD.19,20 GH replacement therapy can result in increased lean body mass and decreased visceral adipose tissue21 and may decrease total and LDLc levels, although effects on HDLc have been inconsistent.22 Endothelial dysfunction improves with GH replacement therapy, with increased flow-mediated dilatation and reduced arterial stiffness due to improved nitric oxide (NO) availability.23 Although GH replacement therapy has been shown to reduce IMT, effects on cardiovascular outcomes are uncertain.24

Figure 1. Effect of growth hormone deficiency on atherosclerosis. GHD, growth hormone deficiency; IGF-1, of insulin-like growth factor type 1; NO, nitric oxide. Adapted with permission from Colao A. 18
Cardiac Structure and functionEchocardiography in patients with childhood- or adolescent-onset GHD has revealed significant reductions in left ventricular (LV) posterior wall thickness and interventricular septal thickness, with resultant decreases in LV mass index and LV internal diameter.25,26 Most adult patients with GHD have impaired LV performance at peak exercise, and report exercise intolerance.27 Several studies have shown that GH replacement therapy improves cardiac performance and increases LV mass, LV end diastolic volume (LVEDV), and stroke volume.25,28
Acromegaly OverviewAcromegaly is characterized by high circulating GH and IGF-1 levels, and is caused by a benign pituitary adenoma in >98% of cases. The morbidity and mortality associated with acromegaly are due to the metabolic effects of GH/IGF-1 hypersecretion and the mass effects of the pituitary adenoma. The mean age at diagnosis is 40-45 years, typically with 5-10 years of symptoms prior to diagnosis. Symptoms include decreased exercise tolerance, increased ring size or ring tightness, increased shoe size, prominence of the jaw and/or forehead, acne or oily skin, arthropathies, and neuropathies.29,30
Diagnosis of acromegaly is suggested by elevated IGF-1 levels, and confirmed by elevated GH levels after administration of an oral glucose tolerance test. Treatments for acromegaly aim to reduce or control adenoma growth, inhibit GH hypersecretion, and normalize IGF-I levels. Surgery is first-line therapy for acromegaly. Treatment options for persistently elevated GH and/or IGF-1 levels include medical therapy and radiotherapy. The three drug classes available for acromegaly treatment are somatostatin analogs, dopamine agonists, and GH receptor antagonists.31
Acromegaly and Cardiovascular Disease Cardiovascular RiskHypertension occurs in 20%-50% of patients with acromegaly. Possible mechanisms include increased arterial stiffness due to hypertrophy and fibrosis of the arterial muscular tunica.32 Acromegaly is also associated with an increased prevalence of diabetes mellitus.33 Systolic and diastolic blood pressure and glycemic control improve with normalization of IGF-1 levels.34
Cardiac Structure and FunctionCardiac histological abnormalities in acromegaly include myocyte hypertrophy, interstitial fibrosis, inflammatory cell infiltration, reduced capillary density, myofibril derangement, and extracellular collagen deposition. The impact of these changes on the structure and function of myocardial and valvular tissues is determined by the duration and severity of GH/IGF-1 excess. In the early stage of acromegaly, there is enhanced myocardial contractility, decreased systemic vascular resistance, increased cardiac output, and overall increased cardiac performance. Relative wall thickness (LV wall thickness/LV radius) increases and causes a reduction in wall stress. In the intermediate stage, after about 5 years of active disease, there is biventricular hypertrophy, diastolic dysfunction, and impaired exertional cardiac performance. Late-stage acromegalic cardiomyopathy is characterized by systolic and diastolic dysfunction, increased myocardial mass, ventricular cavity dilatation, and increased systemic vascular resistance.32 Acromegalic cardiomyopathy is frequently present at diagnosis. Up to two thirds of patients with acromegaly meet echocardiographic criteria for left ventricular hypertrophy (LVH), including about half of all normotensive acromegalics. Patients with severe cardiomyopathy may progress to heart failure, with heart failure seen in 3%-10% of patients.35 Successful treatment of acromegaly halts the progression of cardiac dysfunction, and reduces cardiovascular mortality.29 Surgical cure has been reported to reduce cardiac mass and improve diastolic filling.36 Successful disease control with somatostatin analogs has been shown to improve diastolic filling parameters, reduce volume overload, reduce pulmonary and wedge pressures, and enhance cardiac performance.37 Some evidence suggests that cardiac hypertrophy is reversible in younger patients with a short duration of disease.27 Improvement in LV ejection fraction at peak exercise is also seen in younger patients with short disease duration.38
Cardiac valve disease (aortic and mitral regurgitation) is frequent in acromegaly.39 GH/IGF-1 excess may lead to abnormal extracellular matrix regulation and thus to pathogenesis of myxomatous valvulopathy. The risk of valve disease increases significantly with the duration of GH excess. Aortic and mitral valve dysfunction often persist despite treatment of hormonal excess.40
RhythmElectrocardiogram (ECG) and Holter studies have documented cardiac rhythm abnormalities in acromegaly. Resting ECG changes include left axis deviation, increased QT intervals, septal Q-waves, and ST-T wave depression.41 Additionally, up to 56% of patients with active acromegaly have late potentials on ECG that could predispose to arrhythmias.42 Rhythm disturbances, seen mainly during physical exercise, include atrial and ventricular ectopic beats, paroxysmal atrial fibrillation, paroxysmal supraventricular tachycardia, sick sinus syndrome, bundle branch block, and ventricular tachycardia. The frequency of ventricular premature complexes increases with the duration of acromegaly. The severity of ventricular arrhythmias correlates with increases in LV mass.43 Somatostatin analogs have been shown to reduce QT intervals, and to improve the arrhythmic profile in acromegalic patients.44
Adrenocorticotropic Hormone OverviewAdrenocorticotropic hormone is synthesized and secreted by corticotroph cells of the anterior pituitary gland. The primary role of ACTH is to regulate adrenal cortisol secretion. Excess ACTH can be produced by pituitary corticotroph adenoma or, rarely, by an extrapituitary tumor (ectopic ACTH syndrome) such as small cell lung cancer, carcinoid tumor, or medullary thyroid cancer. This excess ACTH secretion results in hypercortisolism, or Cushing's syndrome. Endogenous Cushing's syndrome is caused by excessive secretion of ACTH (ACTH-dependent cases) in approximately 80% of cases, and by ACTH-independent causes in approximately 20% of cases that include cortisol secretion by unilateral adrenal adenomas, or by bilateral adrenal hyperplasia or dysplasia.45 The overall incidence of endogenous Cushing's syndrome is 2.3 cases per million annually.46
The diagnosis of Cushing's syndrome requires demonstration of elevated cortisol levels with at least two confirmatory tests, including 24-hour urinary-free cortisol, late-night salivary free cortisol, or overnight dexamethasone suppression test. 47,48 The goals of treatment in Cushing's syndrome are normalization and long-term control of cortisol levels, and reversal of clinical features such as weight gain, central obesity, fatigue, muscle weakness, hypertension, diabetes, hirsutism, acne, and menstrual disorders. Treatment options include transsphenoidal surgery, unilateral or bilateral adrenalectomy, radiotherapy and medical therapy. The selection and efficacy of any given treatment modality depends on the underlying cause of hypercortisolism.49 Medical control of hypercortisolism in nonsurgical patients may be achieved using ketoconazole, metyrapone, and/or mitotane.50
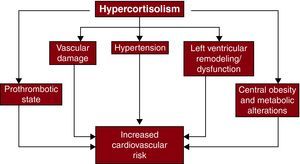
Cushing's Syndrome and Cardiovascular Disease Cardiovascular RiskHypercortisolism is associated with hypertension, central obesity, insulin resistance, dyslipidemia, and alterations in clotting and platelet function51 (Figure 2). Hypertension is present in about 80% of adult patients with endogenous Cushing's syndrome, and results from changes in regulation of plasma volume, systemic vascular resistance, and vasodilatation.53,54 Treatment of Cushing's syndrome usually results in improvement or resolution of hypertension, although hypertension may persist in patients with long-standing hypercortisolism and/or co-existing essential hypertension.55 Abnormal glucose metabolism in Cushing's syndrome results from stimulation of hepatic gluconeogenesis and glycogenolysis. Patients with hypercortisolism may have impaired fasting glucose, impaired glucose tolerance, hyperinsulinemia, insulin resistance, and/or diabetes mellitus.56 Cushing's syndrome has been associated with increased lipoprotein (a), decreased HDLc, and increased triglycerides.54 The duration of cortisol excess correlates with the degree of dyslipidemia seen. Cortisol also increases the synthesis of several coagulation factors, stimulating endothelial production of von Willebrand factor and concomitantly increasing factor VIII.57 Hypercortisolism may also enhance platelet aggregation and reduce plasma fibrinolytic capacity.58,59

Figure 2. Mechanisms of increased cardiovascular risk mediated by hypercortisolism. Reprinted with permission from Fallo et al. 52
Cardiac Structure and FunctionCushing's syndrome has been associated with LVH, concentric remodeling, diastolic dysfunction, and subclinical LV systolic dysfunction.60 Echocardiography has revealed increased interventricular septum thickness and posterior wall thickness, increased LV mass index, and increased relative wall thickness in Cushing's patients. Diastolic dysfunction has been demonstrated, with impaired early LV relaxation, longer isovolumetric relaxation times, and evidence of global myocardial relaxation impairment. The abnormalities of LV structure and function may be reversible with normalization of hypercortisolism. However, patients may continue to exhibit exercise intolerance due to steroid-induced myopathy and resultant muscle weakness.61
Thyroid and the cardiovascular system Thyroid OverviewThyroid dysfunction is common. Hyperthyroidism is present in 1.3% of the United States population (overt in 0.5% and subclinical in 0.7%), and hypothyroidism in 4.6% of the population (overt in 0.3% and subclinical in 4.3%).62 The prevalence of both hypothyroidism and hyperthyroidism increases with age. Data from the Framingham Heart Study have demonstrated suppressed thyrotropin (TSH) levels in 3.9% of patients over age 60 years, and some degree of hypothyroidism, as evidenced by elevated serum TSH levels (>5 mU/L), in 10.3% of unselected patients over age 60 years, with a higher incidence in women (13.6%) than in men (5.7%).63,64
Hyperthyroidism OverviewOvert thyrotoxicosis, or hyperthyroidism, is defined by elevated peripheral free thyroid hormone levels (T3 and/or T4) and a decreased or undetectable TSH. Thyrotoxicosis may result from autoimmune disease, thyroid nodule autonomy, or exogenous thyroid hormone ingestion. Hyperthyroid patients often present with signs and symptoms related to the cardiovascular system including palpitations, sinus tachycardia, atrial fibrillation, systolic hypertension, widened pulse pressure, exercise intolerance, and exertional dyspnea. Other symptoms include fatigue, weight loss, heat intolerance, and diarrhea. Treatments for hyperthyroidism include antithyroid medications (methimazole, carbimazole, and propylthiouracil), beta-blockers, radioactive iodine ablation, and thyroid surgery. Subclinical hyperthyroidism is defined by low or undetectable serum TSH and normal peripheral free thyroid hormone levels. Patients are usually asymptomatic, but remain at risk for some cardiovascular changes associated with hyperthyroidism.65 Consensus panel recommendations suggest consideration of treatment for a persistently suppressed serum TSH (TSH <0.1mIU/L).66
Hyperthyroidism and Cardiovascular Disease HemodynamicsGenomic and nongenomic actions of thyroid hormone result in cardiovascular hemodynamic changes in overt hyperthyroidism that include decreased systemic vascular resistance (SVR), increased heart rate, increased cardiac preload, and increased cardiac output.67,68 SVR is reduced in hyperthyroidism due to thyroid hormone-mediated relaxation of vascular smooth muscle cells and increased endothelial NO production.69,70 The decrease in SVR activates the renin-angiotensin-aldosterone system, leading to increased plasma volume and increased cardiac preload. Thyroid hormone also promotes an increase in blood volume via up-regulation of erythropoietin secretion, further enhancing cardiac preload.71 The combination of increased preload and decreased SVR leads to increased cardiac output.72 Increases in contractility and in resting heart rate further contribute to the increase in cardiac output, which may be 50%-300% higher than normal in overtly hyperthyroid patients.73,74 Treatment of hyperthyroidism reverses these hemodynamic changes.
Cardiovascular RiskSystolic hypertension may be seen in up to 30% of hyperthyroid patients.75 This elevation in systolic pressure may result from the combined effect of increased preload and cardiac output, and decreased arterial compliance.76
Cardiac Structure and FunctionLVH has been associated with hyperthyroidism.77 The hemodynamic changes in hyperthyroidism result in increased cardiac work and compensatory cardiac hypertrophy over time.78 Hyperthyroidism is also associated with enhanced diastolic relaxation. In the short term, hyperthyroidism may be associated with improved diastolic function. However, in the long term, chronic thyrotoxicosis may induce LVH and diastolic dysfunction.79
Exercise intolerance and dyspnea on exertion in overt hyperthyroidism may result from an inability to further increase heart rate and ejection fraction, or to further decrease SVR in the setting of exercise. Hyperthyroid patients may also have skeletal and/or respiratory muscle weakness that further reduces exercise capacity. Patients with subclinical hyperthyroidism may also have decreased exercise tolerance.65 Treatment of hyperthyroidism results in improved exercise tolerance and resolution of exertional dyspnea.80
RhythmSinus tachycardia occurs in approximately 40% of cases of overt hyperthyroidism, and generally resolves after restoration of euthyroidism.81 Subclinical hyperthyroidism is also associated with an increased heart rate.65 Atrial fibrillation is the second most common arrhythmia in overt hyperthyroidism, and occurs in 10%-15% of patients, its prevalence increasing with age.82 Patients with subclinical hyperthyroidism also have an increased risk of atrial fibrillation.65,83 In overtly hyperthyroid patients, factors independently predictive of atrial fibrillation include increasing age, history of cardiac failure, diabetes, elevated systolic or diastolic blood pressure, and LVH on ECG.84 Sinus rhythm can be restored in up to two thirds of patients with overt hyperthyroidism; however, increased age and duration of atrial fibrillation correspond with higher rates of persistent arrhythmia.84 There is limited evidence that treatment of subclinical hyperthyroidism facilitates reversion of atrial fibrillation to normal sinus rhythm.66
Hypothyroidism OverviewHypothyroid patients may present with fatigue, weight gain, cold intolerance, constipation, mild diastolic hypertension, narrowed pulse pressure, and bradycardia. Overt hypothyroidism is characterized by elevated serum TSH and decreased peripheral thyroid hormone levels, with etiologies including autoimmune thyroid gland failure, iatrogenic failure (radioactive iodine, external beam radiation), and thyroidectomy. The treatment of hypothyroidism consists of thyroxine (T4) replacement. Subclinical hypothyroidism is defined by elevated serum TSH with normal peripheral free thyroid hormone levels. Patients with subclinical hypothyroidism are generally asymptomatic or mildly symptomatic. Consensus panel recommendations suggest initiation of thyroid hormone replacement therapy in patients with serum TSH values greater than 10mIU/L, and consideration of replacement therapy in patients with serum TSH 4.5-10mIU/L who have symptoms, and/or high background cardiovascular risk, and/or thyroid autoimmunity.85
Hypothyroidism and Cardiovascular Disease HemodynamicsThe hemodynamic changes in hypothyroidism are the opposite of those seen in hyperthyroidism. Overt hypothyroidism is associated with increased SVR, normal or decreased resting heart rate, decreased contractility, and decreased cardiac output. In addition, diastolic pressure is increased and pulse pressure is narrowed. Cardiac output may be reduced by up to 30%-40% as a result of decreased stroke volume and heart rate.86 The hemodynamic changes of hypothyroidism resolve with restoration of euthyroidism, with normalization of SVR and improved cardiac contractility, and with improved cardiac output.87
Cardiovascular RiskOvert hypothyroidism is associated with accelerated atherosclerosis and coronary artery disease that may be attributable to diastolic hypertension, impaired endothelial function, and hypercholesterolemia. Significant diastolic hypertension may be seen in up to 20% of patients with overt hypothyroidism. This increase in diastolic pressure is the result of increased systemic vascular resistance and increased arterial stiffness, and resolves with T4 replacement therapy.88 Overt hypothyroidism has also been associated with hyperhomocysteinemia, increased C-reactive protein levels, and altered coagulation parameters.88 Subclinical hypothyroidism has been associated with elevated diastolic pressure and increased carotid artery IMT that may improve with T4 replacement.65
Lipid metabolism is altered in hypothyroidism, and approximately 90% of patients with overt hypothyroidism have elevated total cholesterol and LDLc levels.89 Serum total and LDLc levels are increased by approximately 30% in hypothyroidism, with greater increases in LDL levels seen in patients with insulin resistance and in smokers. These increased LDL levels are primarily because of decreased fractional clearance of LDL that results from a reduced number of hepatic LDL receptors. Apolipoprotein B and the atherogenic LDL variant, lipoprotein(a), are also increased in hypothyroidism. Triglyceride and very low density lipoprotein levels are normal to increased, whereas changes in HDL are variable. 90 These lipid abnormalities are generally reversible with restoration of euthyroidism. Subclinical hypothyroidism has been associated with increased LDL and total cholesterol levels in several cross-sectional studies, but the effects of treatment in small trials have been inconsistent.65
Cardiac Structure and FunctionIn hypothyroidism there is resting LV diastolic dysfunction, and both systolic and diastolic dysfunction with exertion. In overt hypothyroidism, impaired LV diastolic function has been demonstrated by slowed myocardial relaxation and impaired early ventricular filling.88 In elderly patients who may have preexisting increased myocardial stiffness, overt hypothyroidism can lead to diastolic heart failure. T4 replacement resolves these functional abnormalities, improving both diastolic and systolic function. Alterations in resting LV diastolic dysfunction have also been demonstrated in patients with subclinical hypothyroidism, with improvements seen in response to T4 replacement.65
Pericardial effusions occur in up to 25% of patients with overt hypothyroidism, and are likely due to increased capillary permeability, increased volume of distribution of albumin, and impaired lymphatic drainage.67 These pericardial effusions accumulate slowly and are seldom hemodynamically significant, although rare cases of cardiac tamponade have been reported.91 Pericardial effusions associated with hypothyroidism generally resolve after 2-3 months of thyroid hormone replacement therapy.67
RhythmECG changes in hypothyroidism include sinus bradycardia, low voltage complexes (small P waves or QRS complexes), prolonged PR and QT intervals, and flattened or inverted T waves.92 Cases of ventricular conduction abnormalities have been reported in association with hypothyroidism, and may be related to QT interval prolongation.73
Amiodarone and Thyroid HormoneAmiodarone, a benzofuranic iodine-rich antiarrhythmic drug, causes thyroid dysfunction in 15%–20% of treated patients, either causing hypothyroidism or thyrotoxicosis. Amiodarone-induced hypothyroidism (AIH) results from persistent iodine-induced inhibition of thyroid gland function, and is more prevalent in patients with preexisting thyroid autoimmunity.93 Treatment of AIH is with T4 replacement. High T4 doses are often required because amiodarone decreases deiodinase activity, resulting in decreased conversion of T4 to the active form, T3. Amiodarone-induced thyrotoxicosis (AIT) is present in two forms: type 1 AIT, or iodine-induced hyperthyroidism, and type 2 AIT, or destructive thyroiditis. Type 1 AIT results in the synthesis and release of excess thyroid hormone, whereas Type 2 AIT results in the release of preformed thyroid hormone from the inflamed thyroid gland. Differentiating between the two forms can be difficult, and management of AIT can be challenging. Type 1 AIT is managed with antithyroid drugs and possibly potassium perchlorate. Type 2 AIT is managed with glucocorticoids, beta-blockade, and rarely thyroidectomy.94 (Table 1) Baseline thyroid function tests and measurements of thyroid peroxidase antibodies should be performed prior to initiating amiodarone, and thyroid function should be monitored every 6 months for the duration of amiodarone therapy.95
Table 1. Features of Amiodarone-Induced Thyroid Dysfunction.
| Type I thyrotoxicosis | Type II thyrotoxicosis | Hypothyroidism | |
| Mechanism | Excess iodine. More common in iodine-deficient areas | Destructive inflammatory thyroiditis | Excess iodine. More common in iodine-sufficient areas |
| Thyroid antibodies | Often present | Usually absent | Often present |
| Thyroid function | Thyrotoxicosis | Thyrotoxicosis | Hypothyroidism |
| 24-h 123Iodine uptake | Usually low in iodine-sufficient regions, but may be normal or increased in iodine-deficient areas | <5% | Usually low in iodine-sufficient regions |
| Color Doppler ultrasound | Hypervascularity | Reduced blood flow | Normal vascularity |
| Therapy | High doses of anti-thyroid drugs; possibly perchlorate or iopanoic acid prior to thyroidectomy | High-dose corticosteroids; Iopanoic acid | Levothyroxine sodium |
Reprinted with permission from Pearce et al. 95
A low serum T3 is the most common thyroid function abnormality in patients with heart failure, and is present in about 10%–30% of patients.96 The biochemical profile of thyroid function in heart failure is consistent with non-thyroidal illness, or euthyroid sick syndrome. It remains unclear whether this reduction in T3 is an adaptive or maladaptive process.97 Additionally, the role of thyroid hormone therapy remains unclear in patients with heart failure and low serum T3 levels. Goals of therapy would include improvements in LV function, remodeling, and microcirculation. Current areas of research include thyroid hormone replacement with T3 and/or T4, use of thyroid hormone analogs (e.g. diiodothyropropionic acid), and gene therapy to modify thyroid hormone receptor or deiodinase expression and activity.98 However, these approaches remain experimental.
Parathyroid hormone and the cardiovascular system Parathyroid Hormone OverviewParathyroid hormone (PTH) plays a critical role in maintaining an adequate calcium–phosphorus homeostasis.99 PTH affects three principal target organs to maintain calcium balance: bone, intestinal mucosa, and kidney. The incidence of primary hyperparathyroidism (PHPT) is approximately 21.6 per 100,000 annually, with a higher incidence in females and in older adults, reaching a peak of 63.2 per 100,000 annually at ages 65-74 years.100 Hypoparathyroidism is much less common.
Hyperparathyroidism OverviewHyperparathyroidism is characterized by inappropriately elevated PTH levels in the setting of elevated calcium concentrations. Causes of hyperparathyroidism include PHPT due to an autonomous adenoma or parathyroid gland hyperplasia, and secondary hyperparathyroidism due to chronic kidney disease or long-standing vitamin D deficiency. The clinical presentation of PHPT has evolved over the past several years as disease detection has improved. Approximately 85% of patients presenting with PHPT are asymptomatic or minimally symptomatic. The diagnosis of hyperparathyroidism is made by measuring serum calcium and serum intact PTH concentrations, and finding inappropriately elevated PTH in the setting of elevated calcium levels. Patients who have symptomatic hyperparathyroidism should be offered surgical management with removal of hyperfunctioning parathyroid adenoma(s). Asymptomatic patients can be clinically monitored or offered surgical management if they meet clinical criteria.101
Hyperparathyroidism and Cardiovascular Disease Cardiovascular RiskThe cardiovascular risk associated with PHPT is attributable in large part to an increased prevalence of hypertension, obesity, glucose intolerance, and insulin resistance.102,103 Proposed mechanisms of hypertension in patients with PHPT include increased calcium deposition leading to arterial stiffness in long standing and/or severe disease, direct PTH-mediated stimulation of the renin-aldosterone system, and PTH-mediated endothelial dysfunction and increased sympathetic activity.104,105 Surgical correction of hyperparathyroidism has not consistently demonstrated improvement in hypertension.106,107 Treatment of PHPT with surgery has been shown to improve insulin sensitivity in patients with more severe disease.108,109 Carotid IMT has been shown to be higher in patients with PHPT, and measures of carotid stiffness are associated with the degree of PTH elevation. This suggests that vessel stiffness may be related to the severity of hyperparathyroidism.110
Cardiac Structure and FunctionLVH has been observed in PHPT in many studies, particularly in patients with moderate to severe hyperparathyroidism, independent of the effects of hypertension. Data from animal studies suggest that PTH has trophic effects on cardiomyoctes that results in hypertrophy. Surgical correction of hyperparathyroidism has resulted in regression of LVH in some studies.111
Diastolic dysfunction has been documented in modest to severe PHPT, with reports of a decreased E/A ratio and prolonged isovolumetric relaxation time. However, it remains unclear whether this effect is attributable more to hypercalcemia or PTH excess.111 Mild PHPT has been inconsistently associated with abnormalities in diastolic dysfunction.
Calcifications of the aortic valve, mitral valve, and myocardium have been demonstrated in PHPT patients with significant hypercalcemia.102 However, studies in patients with mild to moderate hypercalcemia have not demonstrated a consistent correlation with increased valvular calcifications.112
RhythmHypercalcemia, particularly serum calcium >12mg/dL, reduces the plateau phase of the ventricular cardiac action potential and the effective refractory period. ECG findings in significant hypercalcemia include shortened QT and QTc intervals, increased QRS complex amplitude, early peaking and gradual down slope of the descending limb of the T wave, biphasic T waves, and shortened ST segment intervals.92 Successful surgical correction of hyperparathyroidism with reduction in serum calcium concentrations can result in lengthening of the QT and QTc intervals.113 It remains unclear whether hyperparathyroidism and hypercalcemia result in clinically relevant cardiac conduction abnormalities.114,115
Hypoparathyroidism OverviewHypoparathyroidism is characterized by inappropriately low or undetectable PTH levels in the setting of hypocalcemia. Hypoparathyroidism may be congenital or acquired, with surgical removal or damage to the parathyroid glands being the most common acquired cause.116 The signs and symptoms of hypoparathyroidism result from hypocalcemia. Mild hypocalcemia may present with neuromuscular irritability such as perioral numbness, muscle cramping, parethesisas, and positive Chvostek's and Trousseau's signs. Severe hypocalcemia may present with carpopedal spasm, laryngospasm, tetany, and seizures. Diagnostic evaluation should include measurements of serum total and ionized calcium, albumin, phosphorus, magnesium, creatinine, intact PTH, and 25-hydroxyvitamin D.117 Treatment consists of adequate calcium replacement with either oral or IV calcium, and vitamin D metabolites and analogs as needed.
Hypoparathyroidism and cardiovascular disease Cardiac Structure and FunctionThere are case reports of decreased myocardial performance, dilated cardiomyopathy, and congestive heart failure in patients with acute and chronic hypocalcemia.118,119 The mechanism of the myocardial dysfunction is unclear, but may be related to impaired excitation-contraction coupling. Reversal of heart failure and correction of cardiomyopathy have been seen in select cases where correction of calcium deficiency was necessary for clinical and hemodynamic improvement.120,121
RhythmQT prolongation is the ECG hallmark of hypocalcemia, and results from prolongation of the plateau phase of the ventricular cardiac action potential. The rate of change in extracellular calcium levels modulates calcium channel function. Rapid changes in serum calcium results in more marked QT interval changes.122 T-wave changes are not common in hypocalcemia because phase 3 of the action potential is not affected. However, in severe hypocalcemia, T-wave flattening, terminal T-wave inversion, or deeply inverted T waves have been described.92 Hypocalcemia has also rarely been associated with ST segment elevation, possibly due to coronary artery spasm.123
The adrenal gland and the cardiovascular system Aldosterone OverviewAldosterone is a mineralocorticoid hormone produced in the adrenal gland. Aldosterone secretion is regulated primarily by the renin-angiotensin system, although other regulatory factors include serum sodium and potassium levels and ACTH. Mineralocorticoid hormones work to maintain normal sodium and potassium concentrations, and to maintain normal volume status.
Primary Aldosteronism OverviewPrimary aldosteronism (PA), or primary hyperaldosteronism, is a group of conditions in which aldosterone production is inappropriately high, resulting in suppression of the renin-angiotensin system. Hypertension is the clinical hallmark of PA, with the prevalence of PA reported as 0.5%-4.8% of patients with general hypertension, and 4.5%-22% of patients with resistant hypertension.124 Potassium depletion is also characteristic of hyperaldosteronism. The diagnosis of PA is made initially by measuring plasma aldosterone and plasma renin activity, and calculating an aldosterone to renin ratio (ARR). Patients with a positive ARR (ARR >20 with aldosterone >15ng/dL), should undergo confirmatory testing (oral sodium loading, saline infusion, fludrocortisone suppression, or captopril challenge). Common causes of PA include unilateral autonomous adrenal adenoma, and unilateral or bilateral adrenal hyperplasia. A rare cause of PA is a heritable condition known as glucocorticoid-remediable aldosteronism (GRA). Treatment guidelines recommend unilateral laparoscopic adrenalectomy in patients with documented unilateral PA, or medical treatment with a mineralocorticoid receptor antagonist (spironolactone or eplerenone) in nonsurgical patients.125 Medical treatment is suggested for patients with bilateral adrenal disease. Glucocorticoid replacement at the lowest therapeutic dosage is suggested as treatment of GRA.
Primary Aldosteronism and Cardiovascular Disease Cardiovascular RiskPA is associated with hypertension, endovascular dysfunction, and altered glucose metabolism. Mechanisms contributing to hyperaldosteronism-mediated hypertension include plasma volume expansion from sodium and fluid retention, and vasoconstriction from potassium depletion.126 Aldosterone has been shown to decrease NO bioavailability, inhibiting endothelium-dependent relaxation. Aldosterone-mediated perivascular fibrosis reduces vascular compliance.124 Unilateral laparoscopic adrenalectomy in patients with aldosterone-producing adenoma or unilateral adrenal hyperplasia results in normalization of hypokalemia in all patients, improved blood pressure control in nearly all patients, and long-term hypertension cure rates of 30-60%. In PA due to bilateral adrenal disease, unilateral or bilateral adrenalectomy seldom corrects hypertension, necessitating continued mineralocorticoid receptor antagonist therapy.127 Impaired glucose tolerance and decreased insulin sensitivity have been reported in some patients with PA. Proposed mechanisms include direct effects of aldosterone on insulin receptor function, and effects of hypokalemia on insulin regulation.128
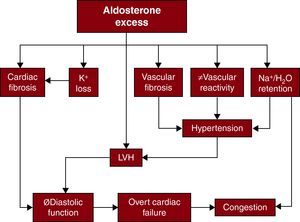
Cardiac Structure and FunctionHyperaldosteronism causes maladaptive cardiac remodeling and has been associated with LVH, cardiac fibrosis, and diastolic dysfunction.129,130 (Figure 3)131 The degree of LVH seen in PA exceeds the effects of hypertension alone.132 In animal models, aldosterone has been shown to directly stimulate cell growth and cardiomyocyte hypertrophy.133 Aldosterone has also been shown to promote collagen deposition, activation of inflammatory cells, and stimulation of fibroblast proliferation.134,135 Diastolic dysfunction has been demonstrated with lower early/late-wave diastolic filling velocities ratio, and longer deceleration time in patients with PA.136 Surgical and medical treatments may be effective in reducing LV mass, with decreases in blood pressure and plasma aldosterone levels predictive of response to therapy.

Figure 3. Mechanisms by which aldosterone excess may bring about adverse cardiovascular sequelae. LVH, left ventricular hypertrophy. Adapted with permission from Stowasser. 131
Congestive Heart Failure and Aldosterone BlockadeIn conditions such as heart failure and myocardial infarction, aldosterone levels are elevated and contribute to pathologic cardiovascular remodeling via direct effects on collagen deposition and resultant cardiovascular fibrosis.137 Elevated aldosterone levels also promote endothelial dysfunction and vascular inflammation. Clinical studies have shown that aldosterone blockade reduces LV remodeling and collagen deposition, improves endothelial function, decreases inflammation, and increases myocardial perfusion.138,139,140 Following two landmark randomized controlled trials, the Randomized Aldactone Evaluation Study (RALES) and the Eplerenone Postacute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS), aldosterone blockade was added to clinical guidelines for management of chronic heart failure.141,142 The addition of an aldosterone antagonist is recommended in selected patients with moderately severe to severe symptoms of heart failure and reduced LVEF, or with LV dysfunction early after myocardial infarction, who can be carefully monitored for preserved renal function and normal potassium concentration.143 The effectiveness of aldosterone blockade in diastolic dysfunction and in mild-to-moderate heart failure is unclear.140,144
Pheochromocytoma OverviewPheochromoctyomas are catecholamine-producing tumors that originate from chromaffin cells of the adrenal medulla and the sympathetic ganglia (catecholamine-secereting paragangliomas, or extra-adrenal pheochromocytomas). The estimated prevalence of pheochromocytoma is 0.05-0.12% of the general population, and 0.2-0.6% of patients with hypertension.145 Patients may present asymptomatically if diagnosed after detection by adrenal imaging or genetic testing. Symptomatic patients present with hypertension (episodic or sustained) and paroxysmal symptoms such as dizziness, headache, flushing, diaphoresis, and palpitations. The diagnosis of pheochromocytoma is made with biochemical confirmation of catecholamine excess, utilizing urinary and plasma measurements of metanephrines and catecholamines, followed by radiologic evaluation for tumor localization. Treatment of pheochromocytoma consists of surgical resection, with preoperative medical optimization to obtain adequate blood pressure control and volume expansion.146,147
Pheochromocytoma and Cardiovascular Disease Cardiovacular RiskHypertension is present in over 50% of patients with pheochromocytoma, and may be sustained or paroxysmal. Higher variability of blood pressure has been demonstrated in pheochromocytoma compared to patients with essential hypertension, and is associated with a higher incidence of target organ damage.148 Resolution of hypertension has been reported in about 50% of patients after successful surgical treatment of pheochromocytoma.149
Markers of endothelial dysfunction, such as increased carotid IMT, have been demonstrated in patients with pheochromocytoma.150 These changes have been attributed to the effects of excess catecholamines on vascular wall growth and thickening. Normalization of catecholamine levels after surgical removal of pheochromocytoma has been shown to improve carotid IMT, and reduce carotid wall fibrosis.151
Cardiac Structure and FunctionExcess catecholamine action in pheochromocytoma can lead to cardiomyopathy, ischemic heart disease, myocardial stunning, and, rarely, cardiogenic shock. The incidence of cardiomyopathy in patients with pheochromocytoma is about 26%, with primary manifestations including dilated cardiomyopathy and hypertrophic cardiomyopathy.152 Echocardiogram may reveal LV dilatation with diffuse decrease in contractility, left atrial dilatation with increased end-diastolic pressure, reduced ejection fraction, and septal hypertrophy. In the setting of intravascular volume depletion and impaired diastolic filling, patients may present with an outflow obstruction that mimics hypertrophic obstructive cardiomyopathy. Frequently seen on echocardiogram, LVH is attributable more to hypertension than to catecholamine effects.150
Patients with pheochromocytoma-associated cardiomyopathy may present with pulmonary edema, or with acute chest pain and myocardial ischemia/infarction. Pulmonary edema results from increased pulmonary capillary permeability, increased peripheral vascular resistance, increased hydrostatic pressure, and overfilling or constriction of efferent pulmonary veins. Myocardial ischemia or infarction may result from coronary vasospasm, with catecholamine action leading to vasoconstriction, decreased coronary blood flow, and increased cardiac oxygen demand. Myocardial stunning following catecholamine-induced vasospasm has been reported, in addition to case reports of tako-tsubo-like apical dyskinesia leading to acute cardiogenic shock.153
Catecholamine-induced cardiomyopathy has been shown to improve after surgical treatment of pheochromocytoma. Reversal of cardiomyopathy depends on early identification and treatment. The prognosis for patients with acute heart failure and significant myocardial damage is very poor.
RhythmThe electrocardiographic signs related to pheochromocytoma include right-axis deviation, poor R-wave progression, inverted T waves, and QT prolongation. If there is permanent myocardial damage and development of cardiomyopathy, signs of ventricular hypertrophy and ischemia may be present on electrocardiogram. Cardiac arrhythmias may be seen in 20% of patients with pheochromocytoma, and include sinus tachycardia, sick sinus syndrome, supraventricular and ventricular tachycardia.150,152
ConclusionsEndocrine dysfunction may have a significant impact on the cardiovascular system. Restoration of normal endocrine function often results in reversal of adverse cardiovascular changes. Hormone-mediated cardiac changes should be considered when evaluating endocrine and cardiac patients.
Conflicts of interestNone declared.
Corresponding author: 88 East Newton Street, Evans 201, Boston, MA 02118, USA. elizabeth.pearce@bmc.org