The development of pulmonary arterial hypertension (PAH) associated with congenital heart disease (CHD) is a multifactorial process still under research.1 In certain kinds of CHD, vascular remodeling leading to pressure and volume overloads is often a satisfactory explanation, particularly in posttricuspid Eisenmenger syndrome. However, in a high percentage of patients included in the other 3 groups of PAH-CHD, we suspect an unknown individual predisposition, in which CHD acts as a mechanism required for the development of PAH. The best example are patients with small or incidental shunts, as the resulting hemodynamic abnormality itself is insufficient to induce structural changes in the pulmonary vascular tree. These defects also have the potential to act as a relief valve for the right ventricle in severe PAH, delaying the appearance of symptoms such as syncope or those related to heart failure. In patients with PAH associated with systemic-to-pulmonary shunt, the main issue is to identify which patients may undergo successful correction to reverse the process and which patients will experience the appearance of PAH due to closure or postrepair reclassification, with poorer prognosis.2 Postrepair PAH is more likely when correction is undertaken later and the pre-established vascular lesion is larger. However, this subgroup is also identified in patients who undergo early repair, who probably have an individual susceptibility resulting from different factors, in which genetics could be key.
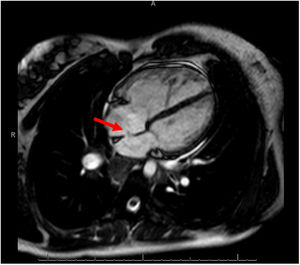
We describe a family with 2 individuals with PAH-CHD who had a variant not previously reported that produced a unique phenotype: different septal defects, hemoptysis, and early onset. The proband is a man diagnosed at age 19 years with PAH plus a muscular ventricular septal defect (VSD) of 6mm, classified as incidental and with predominantly right-to-left shunt, detected after an episode of massive hemoptysis. At onset, multiple bronchial collaterals were found to have developed, and the hemodynamic study confirmed suprasystemic pulmonary pressures. Although triple vasodilator treatment with high-dose systemic prostacyclins was introduced, the patient remained in a high-risk situation and finally, 4 years after diagnosis, required a bilateral lung transplant in which a decision was made to keep the VSD open. Ten years after the transplant, the patient was still clinically well. The second patient is the sister of the index case, diagnosed at age 26 years with severe PAH plus a 12-mm ostium secundum atrial septal defect and predominantly left-to-right shunt (figure 1). The diagnosis was also performed after an episode of hemoptysis, in this case, minor. The patient started oral combination therapy, and 4 years after diagnosis was still at low risk.
. Right atrial and ventricular dilatation was also present.")
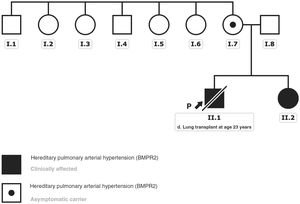
A gene panel for PAH was offered to the patients to determine the etiology. Both patients, and later their family members, gave written consent for the study and for disclosure in a scientific article. The analysis detected a pathogenic variant in the gene that encodes bone morphogenetic protein receptor type 2 (BMPR2): NM_001204.6: c.2674delG: p.(Glu892Asnfs*4). No other candidate variant was identified among the rest of the genes included in the panel designed by our group.3 The subsequent familial study revealed that the variant came from the mother, who remained asymptomatic with no evidence of pulmonary hypertension or septal defects. This mutation was not identified when tracing the mother's siblings and could not be studied in the grandparents, already deceased at the time of the study due to other causes; hence, it was considered an apparently de novo variant (figure 2). Although this variant was also not listed in the population databases consulted (gnomAD genomes, gnomAD exomes, 1000G, ESP, Kaviar, Beacon, Braco), most in silico pathogenicity predictors applied indicate a deleterious effect of the variant, as it generates a premature stop codon.

Genealogical tree of the affected family. The black arrow indicates the index case. Clinically affected patients with the described BMPR2 gene variant are marked in black. The black dot indicates the clinically asymptomatic carrier patient. This figure is shown in full color only in the electronic version of the article.
Our series describes 2 cases of hereditary PAH in which an incidental septal defect could have accelerated the development of pulmonary vascular diseases as a facilitating factor. Regarding genetic susceptibility for the development of PAH in patients with CHD, the prevalence of pathogenic variants of the BMPR2 gene has been reported as being up to 7.5% in patients with PAH related to simple CHD (atrial septal defect, VSD, and persistent ductus arteriosus) compared with 1.2% of patients with CHD without PAH.4 Experimental studies have found a relationship between BMPR2-transforming growth factor-beta (TGF-ß) and the appearance of simple septal defects and even complex septal abnormalities.5 This metabolic pathway is involved in regulating the growth, differentiation, and apoptosis of mesenchymal and epithelial cells, and is related to both the appearance of adverse vascular remodeling and abnormal heart development. Moreover, BMPR2 mutations are the most common in hereditary PAH, with an autosomal dominant inheritance pattern, variable penetrance, and greater expressivity in women. In addition, these patients are usually in a more serious hemodynamic condition at diagnosis and have greater hypertrophy of the bronchial arteries, which are factors related to the appearance of hemoptysis.6 Other genes, such as TBX4 and SOX17 have also been associated with the development of PAH related to CHD.1
In summary, this family highlights the importance of a genetic study in patients with PAH and simple septal defects, which could be extrapolated to the various forms of PAH-CHD. In patients with PAH and incidental shunts and in patients with postrepair PAH, a genetic study may help determine the approach to the disease course and improve treatment and familial assessment. In patients with PAH associated with a large systemic-to-pulmonary shunt, the genetic findings may guide the decision regarding surgical repair of the defect. The 2 patients we describe had incidental defects, and the decision not to perform surgery was made independently from the genetic study. However, because the defects were known, it was possible to predict the clinical progression of the disease and to design a particularly strong drug therapy and an accurate familial assessment.
FUNDINGThis article was funded by the PI 18/01233 project “Genetic-Molecular Basis for Precision Medicine in Pulmonary Arterial Hypertension”, Ministry of Economy and Competitiveness of the Government of Spain (Carlos III Health Institute). A. Cruz Utrilla receives funding through a Rio Hortega agreement (CM20/00164), Ministry of Science and Innovation of Spain (Carlos III Health Institute).
AUTHORŚ CONTRIBUTIONSA. Cruz Utrilla was responsible for the design, writing, and critical analysis of the manuscript, as well as for including the patients in the study. N. Gallego collaborated in the genetic studies and in writing. T. Segura de la Cal collaborated in writing and critical analysis. J. Tenorio-Castaño collaborated in performing the genetic studies and in the critical analysis. F. Arribas-Ynsaurriaga collaborated in the critical analysis. P. Escribano Subias collaborated in the study design, writing, and critical analysis.
CONFLICTS OF INTERESTThe authors declared no conflicts of interests in relation to this study.