ISSN: 0300-8932
Factor de impacto 2023
7,2
SEC 2022 - El Congreso de la Salud Cardiovascular

Palma de Mallorca y online,
20 - 22 de Octubre de 2022
Introducción
Dr. Juan José Gómez Doblas
Presidente del Comité Científico del Congreso
Comité ejecutivo
Comité de evaluadores
Listado de sesiones
Índice de autores
6055. Cardiopatías familiares: miscelánea
Fecha
: 22-10-2022 13:45:00
Tipo
: Pósteres
Sala
: E-poster 3 (Planta 0)
6055-2. KCNH2 P. GLY262ALAFSTER98: UNA NUEVA VARIANTE ASOCIADA AL SÍNDROME DE QT LARGO EN UNA COHORTE ESPAÑOLA
Alejandro Junco Vicente, Alicia Pérez Pérez, Elías Cuesta Llavona, Miguel Soroa Ortuño, Noemi Barja González, Yván Rafael Persia Paulino, José Manuel Rubín López, José Julián Rodríguez Reguero, Bárbara C. Fernández Barrio, Eliecer Coto García, César Morís de la Tassa, Juan Gómez de Oña y Rebeca Lorca Gutiérrez
Hospital Universitario Central de Asturias, Oviedo (Asturias).
Hospital Universitario Central de Asturias, Oviedo (Asturias).
Introducción y objetivos: El síndrome de QT largo (SQTL) es un trastorno arritmogénico hereditario (canalopatía), caracterizado por una repolarización ventricular (intervalo QTc) prolongada asociado con una mayor susceptibilidad a eventos arrítmicos potencialmente mortales. La base patogénica de esta enfermedad implica alteraciones en el funcionamiento de los canales iónicos. El gen KCNH2 codifica la subunidad-α formadora de poros del canal de potasio (Kv11,1) que juega un papel esencial en la repolarización. Las variantes de pérdida de función en KCNH2 pueden alterar el canal de potasio, aumentando la duración del potencial de acción ventricular y, por lo tanto, generando el SQTL. Sigue existiendo gran dificultad para establecer relaciones estrechas genotipo-fenotipo-alto riesgo arrítmico. La descripción de más series resulta fundamental para el conocimiento de esta enfermedad.
Métodos: Realizamos un estudio retrospectivo, revisando todos los casos de SQTL referidos a estudio genético en nuestro centro, localizando los portadores de KCNH2 p. Gly262AlafsTer98 (c.785 delG: NM_000238). El cribado se realizó con muestras de ADN secuenciado por NGS. Se realizaron estudios clínicos y genéticos en todos los familiares disponibles de los casos hallados.
Resultados: Se identificaron tres casos índice portadores de esta nueva variante patógenica no descrita previamente. Analizamos 34 familiares en 3 familias de etnia gitana (figura). En la cohorte se registraron 7 muertes súbitas (4 varones y 3 mujeres) a una edad media 35,6 +/-13,1 años, todas en reposo y acontecidas antes de consultar con cardiología (sin genética ni ECG), dos en portadores obligados. Se indicó desde el diagnóstico tratamiento con β-bloqueantes con una baja adherencia terapéutica. Durante el seguimiento, ningún otro pariente ha sufrido un desenlace fatal. Se realizó estudio genético en 28, siendo 22 portadores, todos los estudiados con fenotipo positivo (ECG con QTc prolongado, corregido por la fórmula de Bazett). 2 pacientes rechazaron evaluación. 1 de cada 4 pacientes había presentado síncope y 3 debutaron con parada cardiaca. 6 pacientes son portadores de DAI, 2 tras parada cardiaca resucitada y 4 con descargas apropiadas en el seguimiento.
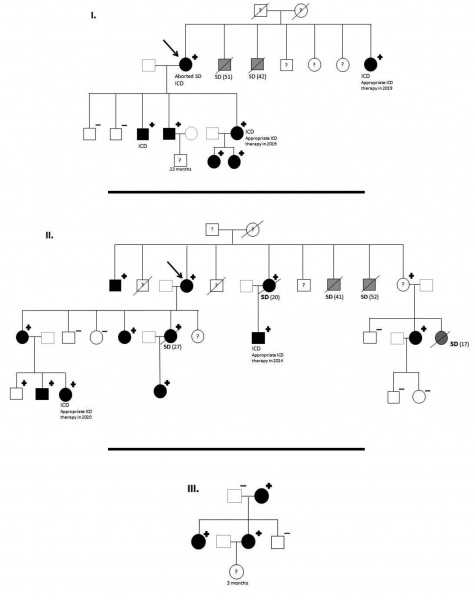
Genealogía: portadores de KCNH2 p. Gly262AlafsTer98.
Conclusiones: Presentamos una nueva variante en KCNH2 asociada a SQTL con alta penetrancia y alto riesgo arrítmico en la juventud, en una población española de especial interés por su baja adherencia a tratamiento médico.
Comunicaciones disponibles de "Cardiopatías familiares: miscelánea"
- 6055-1. MODERADORA
Helena Llamas Gómez, Sevilla (Córdoba)
- 6055-2. KCNH2 P. GLY262ALAFSTER98: UNA NUEVA VARIANTE ASOCIADA AL SÍNDROME DE QT LARGO EN UNA COHORTE ESPAÑOLA
- Alejandro Junco Vicente, Alicia Pérez Pérez, Elías Cuesta Llavona, Miguel Soroa Ortuño, Noemi Barja González, Yván Rafael Persia Paulino, José Manuel Rubín López, José Julián Rodríguez Reguero, Bárbara C. Fernández Barrio, Eliecer Coto García, César Morís de la Tassa, Juan Gómez de Oña y Rebeca Lorca Gutiérrez
Hospital Universitario Central de Asturias, Oviedo (Asturias).
- 6055-3. MANIOBRAS ELECTROCARDIOGRÁFICAS EN EL DESPISTAJE DE CANALOPATÍAS: ¿CUÁL ES LA RENTABILIDAD DIAGNÓSTICA?
- María Maeve Soto Pérez1, Jesús Piqueras Flores2, Jorge Martínez del Río1, Pedro Pérez Díaz3, María Aránzazu González Marín4, Martín Negreira Caamaño1, Cristina Mateo Gómez1, Daniel Águila Gordo1, Andrez Felipe Cubides Novoa1, Pablo Soto Martín1, Emilio Blanco López1, José María Arizón Muñoz1 y Javier Jiménez Díaz5
1Servicio de Cardiología. Hospital General Universitario de Ciudad Real, 2Unidad de Cardiopatías Familiares. Servicio de Cardiología. Hospital General Universitario de Ciudad Real, 3Hospital Infanta Cristina, Parla, Madrid, 4Servicio de Pediatría. Hospital General Universitario de Ciudad Real y 5Unidad de arritmias y electrofisiología. Servicio de Cardiología. Hospital General Universitario de Ciudad Real.
- 6055-4. CARACTERÍSTICAS CLÍNICAS DEL SÍNDROME DE QT LARGO INDUCIDO POR FÁRMACOS EN UNA GRAN COHORTE PROSPECTIVA
- Bieito Campos García, Héctor Hernández Ontiveros, Aina Ávila Parcet, Víctor García Hernando, Mar Carceller Sindreu, Juliana Salazar Blanco, Benjamín Rodríguez Santiago, Ana Juanes Borrego, Concepción Alonso Martín, Enrique Rodríguez Font, Zoraida L. Moreno Weidmann, Francisco Javier Méndez Zurita, Xavier Viñolas Prat y José M. Guerra Ramos
Hospital de la Santa Creu i Sant Pau, Barcelona.
- 6055-5. ESTUDIO DE LAS ALTERACIONES GENÉTICAS EN PACIENTES CON HIPERCOLESTEROLEMIA FAMILIAR DEL ÁREA SANITARIA DE TOLEDO, DETECTADOS MEDIANTE UNA NUEVA ESTRATEGIA DE CRIBADO SISTEMÁTICO PARTIENDO DE ANALÍTICA CENTRALIZADA PREEXISTENTE
- Joaquín Sánchez-Prieto Castillo, Esther Gigante Miravalles, Fernando Sabatel López, Alejandro Cabello Rodríguez, Carlos de Cabo Porras y Luis Rodríguez Padial
Hospital General Universitario de Toledo.
- 6055-6. GRADO DE CONTROL LIPÍDICO DE LOS PACIENTES CON HIPERCOLESTEROLEMIA FAMILIAR EN EL EMPORDÀ
- Patricia García Morante1, Daniel Vidal Soto1, Estel Pons Viñas1, Anna C. Comas Aleix2, Armand Grau Martín1 y Àlex Vila Belmonte1
1Hospital de Figueres, Girona y 2Fundació Salut Empordà, Figueres, Girona.
- 6055-7. PREDICTORES DE ENFERMEDAD CARDIOVASCULAR EN UNA UNIDAD DE HIPERCOLESTEROLEMIA FAMILIAR
- Gustavo Aníbal Cortez Quiroga, María Jesús Huertas Escribano, Elisa Martínez Perona, María de la Paz Eliche Mozas, Ana Cubillas Quero y Lara Cruz Romero
Hospital Alto Guadalquivir, Andújar (Jaén).
- 6055-8. DELECIÓN EN NOTCH 1, NUEVA CAUSA DE ANEURISMA AÓRTICO CON VÁLVULA AÓRTICA TRICÚSPIDE
- Yolanda Rico Ramírez1, Laura Torres-Juan1, Francisca Ramis Barceló2, Jaume Pons Llinares1, Elena Fortuny Frau1, Rafael Félix Ramis1, Víctor J. Asensio1, Icíar Martínez1, Vicente Peral Disdier1 y Damián Heine-Suñer1
1Hospital Son Espases, Palma de Mallorca y 2Clínica Planas, Palma de Mallorca.
- 6055-9. ANEURISMA DE AORTA ASCENDENTE GIGANTE COMO FORMA DE PRESENTACIÓN DE CUTIS LAXA 1B EN PACIENTES JÓVENES: NUEVO CASO Y REVISIÓN BIBLIOGRÁFICA
- Alejandro Used Gavín, José María Larrañaga Moreira, Esteban Martín Álvarez, Borja Souto Caínzos, Carmen Iglesias Gil, Víctor X. Mosquera Rodríguez, Roberto Barriales Villa y José Manuel Vázquez Rodríguez
Complexo Hospitalario Universitario A Coruña.
- 6055-10. ANÁLISIS DE LAS CARACTERÍSTICAS POBLACIONALES Y RENTABILIDAD DE LA IMPLANTACIÓN DE DAI EN PACIENTES CON MIOCARDIOPATÍA DILATADA NO ISQUÉMICA
- María Maeve Soto Pérez1, Jesús Piqueras Flores2, Javier Jiménez Díaz3, Felipe Higuera Sobrino3, Natalia Arance Romero4, Jorge Martínez del Río1, Martín Negreira Caamaño1, Daniel Águila Gordo1, Cristina Mateo Gómez1, Andrez Felipe Cubides Novoa1, Alfonso Morón Alguacil1, Pablo Soto Martín1, Emilio Blanco López1, Daniel Salas Bravo1 y José María Arizón Muñoz1
1Servicio de Cardiología. Hospital General Universitario de Ciudad Real, 2Unidad de Cardiopatías Familiares. Servicio de Cardiología. Hospital General Universitario de Ciudad Real, 3Unidad de Arritmias y Electrofisiología. Servicio de Cardiología. Hospital General Universitario de Ciudad Real y 4Universidad de Castilla-La Mancha, Ciudad Real.
- 6055-11. RELEVANCIA DE LA TAQUICARDIA VENTRICULAR NO SOSTENIDA EN LA MIOCARDIOPATÍA HIPERTRÓFICA
- Charlotte Boillot, Belén Santos González, Ana Díaz Rojo, Andrea González Pigorini, Alejandro Cabello Rodríguez, Álvaro Serrano Blanco y Esther Gigante Miravalles
Complejo Hospitalario de Toledo, SESCAM.
Más comunicaciones de los autores
-
Barja González, Noemi
- 6055-2 - KCNH2 P. GLY262ALAFSTER98: UNA NUEVA VARIANTE ASOCIADA AL SÍNDROME DE QT LARGO EN UNA COHORTE ESPAÑOLA
- 6044-10 - IMPLANTE DE VÁLVULA AÓRTICA TRANSCATÉTER CON TÉCNICA DE SUPERPOSICIÓN DE SENOS CORONARIOS FRENTE A LA TÉCNICA TRADICIONAL PARA REDUCCIÓN DE LA INCIDENCIA DEL BLOQUEO INTERAURICULAR RELACIONADA AL PROCEDIMIENTO: RESULTADOS DE UN ESTUDIO OBSERVACIONAL PROSPECTIVO EMPAREJADO POR PROPENSIÓN
- Coto García, Eliecer
-
Cuesta Llavona, Elías
- 5027-2 - CORRELACIÓN DE LA MICROBIOTA INTESTINAL Y LOS PARÁMETROS NUTRICIONALES EN PACIENTES CON INSUFICIENCIA CARDIACA
- 6055-2 - KCNH2 P. GLY262ALAFSTER98: UNA NUEVA VARIANTE ASOCIADA AL SÍNDROME DE QT LARGO EN UNA COHORTE ESPAÑOLA
- 5027-4 - DIFERENCIAS DE LA MICROBIOTA INTESTINAL RELACIONADAS CON EL PRONÓSTICO DE LOS PACIENTES CON INSUFICIENCIA CARDIACA SEGÚN LA FRACCIÓN DE EYECCIÓN
- Fernández Barrio, Bárbara C.
-
Gómez de Oña, Juan
- 5027-2 - CORRELACIÓN DE LA MICROBIOTA INTESTINAL Y LOS PARÁMETROS NUTRICIONALES EN PACIENTES CON INSUFICIENCIA CARDIACA
- 6055-2 - KCNH2 P. GLY262ALAFSTER98: UNA NUEVA VARIANTE ASOCIADA AL SÍNDROME DE QT LARGO EN UNA COHORTE ESPAÑOLA
- 5027-4 - DIFERENCIAS DE LA MICROBIOTA INTESTINAL RELACIONADAS CON EL PRONÓSTICO DE LOS PACIENTES CON INSUFICIENCIA CARDIACA SEGÚN LA FRACCIÓN DE EYECCIÓN
-
Junco Vicente, Alejandro
- 6049-8 - TRAYECTOS INTRAMIOCÁRDICOS CORONARIOS: PREVALENCIA, CARACTERIZACIÓN, ASOCIACIÓN CLÍNICA Y PRONÓSTICA DE UNA COHORTE DE ANGIOTC CORONARIO
- 6044-10 - IMPLANTE DE VÁLVULA AÓRTICA TRANSCATÉTER CON TÉCNICA DE SUPERPOSICIÓN DE SENOS CORONARIOS FRENTE A LA TÉCNICA TRADICIONAL PARA REDUCCIÓN DE LA INCIDENCIA DEL BLOQUEO INTERAURICULAR RELACIONADA AL PROCEDIMIENTO: RESULTADOS DE UN ESTUDIO OBSERVACIONAL PROSPECTIVO EMPAREJADO POR PROPENSIÓN
- 5031-7 - GEMELOS EN LA MIOCARDIOPATÍA HIPERTRÓFICA. FENOTIPOS OPUESTOS PESE A GENÉTICA IDÉNTICA Y AMBIENTE SIMILAR
- 6049-14 - PREVALENCIA Y CARACTERIZACIÓN DE LAS ANOMALÍAS EN EL ORIGEN AÓRTICO DE LAS ARTERIAS CORONARIAS ESTUDIADAS MEDIANTE TOMOGRAFÍA COMPUTARIZADA
- 6055-2 - KCNH2 P. GLY262ALAFSTER98: UNA NUEVA VARIANTE ASOCIADA AL SÍNDROME DE QT LARGO EN UNA COHORTE ESPAÑOLA
-
Lorca Gutiérrez, Rebeca
- 6055-2 - KCNH2 P. GLY262ALAFSTER98: UNA NUEVA VARIANTE ASOCIADA AL SÍNDROME DE QT LARGO EN UNA COHORTE ESPAÑOLA
- 6038-7 - INFLUENCIA DE LA GENÉTICA EN EL FENOTIPO DE LA MIOCARDIOPATÍA HIPERTRÓFICA
- 5031-7 - GEMELOS EN LA MIOCARDIOPATÍA HIPERTRÓFICA. FENOTIPOS OPUESTOS PESE A GENÉTICA IDÉNTICA Y AMBIENTE SIMILAR
-
Morís de la Tassa, César
- 6049-14 - PREVALENCIA Y CARACTERIZACIÓN DE LAS ANOMALÍAS EN EL ORIGEN AÓRTICO DE LAS ARTERIAS CORONARIAS ESTUDIADAS MEDIANTE TOMOGRAFÍA COMPUTARIZADA
- 6054-5 - CREACIÓN DE UNA UNIDAD MULTIDISCIPLINAR DE CARDIOGERIATRÍA PARA EL MANEJO DE LA INSUFICIENCIA CARDIACA DEL PACIENTE MAYOR
- 6006-10 - ESTRATEGIA DE INMUNOSUPRESIÓN EN TRASPLANTE CARDIACO: PROTECCIÓN FRENTE A CITOMEGALOVIRUS EN ESTRATEGIAS QUE INCLUYEN EVEROLIMUS. DATOS DE DOS CENTROS ESPAÑOLES
- 6022-5 - IMPLANTE DE VÁLVULA AÓRTICA TRANSCATÉTER INFLABLE CON TÉCNICA DE SUPERPOSICIÓN DE SENOS CORONARIOS FRENTE A LA TÉCNICA TRADICIONAL: ALTERACIONES ELECTROCARDIOGRÁFICAS SECUNDARIAS INMEDIATAS Y A LARGO PLAZO
- 4025-4 - REMODELADO ESTRUCTURAL A CORTO PLAZO TRAS IMPLANTE DE TRICVALVE
- 5024-5 - REDUCCIÓN DE LA HOSPITALIZACIÓN CARDIOVASCULAR Y POR CUALQUIER CAUSA TRAS IMPLANTE DE VÁLVULA AÓRTICA TRANSCATÉTER MEDIANTE TÉCNICA DE SUPERPOSICIÓN DE SENOS CORONARIOS: RESULTADOS TRAS UN AÑO DE SEGUIMIENTO
- 5014-7 - DETERIORO COGNITIVO EN EL PACIENTE MAYOR CON ESTENOSIS AÓRTICA GRAVE SINTOMÁTICA: INFLUENCIA EN LA TOMA DE DECISIONES TERAPÉUTICAS E IMPACTO EN LA MORTALIDAD A LOS 12 MESES
- 6022-2 - BÚSQUEDA DE PREDICTORES DE FUTILIDAD EN PACIENTES MAYORES SOMETIDOS A REEMPLAZO VALVULAR AÓRTICO TRANSCATÉTER
- 6044-10 - IMPLANTE DE VÁLVULA AÓRTICA TRANSCATÉTER CON TÉCNICA DE SUPERPOSICIÓN DE SENOS CORONARIOS FRENTE A LA TÉCNICA TRADICIONAL PARA REDUCCIÓN DE LA INCIDENCIA DEL BLOQUEO INTERAURICULAR RELACIONADA AL PROCEDIMIENTO: RESULTADOS DE UN ESTUDIO OBSERVACIONAL PROSPECTIVO EMPAREJADO POR PROPENSIÓN
- 5001-11 - REGISTRO MULTICÉNTRICO DE LITOTRICIA CORONARIA EN EL MUNDO REAL: SEGUIMIENTO CLÍNICO A LARGO PLAZO
- 5024-6 - REDUCCIÓN DE LA TASA DE IMPLANTE DE MARCAPASOS EN TAVI UTILIZANDO LA SUPERPOSICIÓN DEL SENO IZQUIERDO Y DERECHO
- 5031-7 - GEMELOS EN LA MIOCARDIOPATÍA HIPERTRÓFICA. FENOTIPOS OPUESTOS PESE A GENÉTICA IDÉNTICA Y AMBIENTE SIMILAR
- 6055-2 - KCNH2 P. GLY262ALAFSTER98: UNA NUEVA VARIANTE ASOCIADA AL SÍNDROME DE QT LARGO EN UNA COHORTE ESPAÑOLA
- Pérez Pérez, Alicia
-
Persia Paulino, Yván Rafael
- 5024-5 - REDUCCIÓN DE LA HOSPITALIZACIÓN CARDIOVASCULAR Y POR CUALQUIER CAUSA TRAS IMPLANTE DE VÁLVULA AÓRTICA TRANSCATÉTER MEDIANTE TÉCNICA DE SUPERPOSICIÓN DE SENOS CORONARIOS: RESULTADOS TRAS UN AÑO DE SEGUIMIENTO
- 6044-10 - IMPLANTE DE VÁLVULA AÓRTICA TRANSCATÉTER CON TÉCNICA DE SUPERPOSICIÓN DE SENOS CORONARIOS FRENTE A LA TÉCNICA TRADICIONAL PARA REDUCCIÓN DE LA INCIDENCIA DEL BLOQUEO INTERAURICULAR RELACIONADA AL PROCEDIMIENTO: RESULTADOS DE UN ESTUDIO OBSERVACIONAL PROSPECTIVO EMPAREJADO POR PROPENSIÓN
- 6055-2 - KCNH2 P. GLY262ALAFSTER98: UNA NUEVA VARIANTE ASOCIADA AL SÍNDROME DE QT LARGO EN UNA COHORTE ESPAÑOLA
- 6022-5 - IMPLANTE DE VÁLVULA AÓRTICA TRANSCATÉTER INFLABLE CON TÉCNICA DE SUPERPOSICIÓN DE SENOS CORONARIOS FRENTE A LA TÉCNICA TRADICIONAL: ALTERACIONES ELECTROCARDIOGRÁFICAS SECUNDARIAS INMEDIATAS Y A LARGO PLAZO
-
Rodríguez Reguero, José Julián
- 5031-7 - GEMELOS EN LA MIOCARDIOPATÍA HIPERTRÓFICA. FENOTIPOS OPUESTOS PESE A GENÉTICA IDÉNTICA Y AMBIENTE SIMILAR
- 6038-7 - INFLUENCIA DE LA GENÉTICA EN EL FENOTIPO DE LA MIOCARDIOPATÍA HIPERTRÓFICA
- 6055-2 - KCNH2 P. GLY262ALAFSTER98: UNA NUEVA VARIANTE ASOCIADA AL SÍNDROME DE QT LARGO EN UNA COHORTE ESPAÑOLA
-
Rubín López, José Manuel
- 6055-2 - KCNH2 P. GLY262ALAFSTER98: UNA NUEVA VARIANTE ASOCIADA AL SÍNDROME DE QT LARGO EN UNA COHORTE ESPAÑOLA
- 6044-10 - IMPLANTE DE VÁLVULA AÓRTICA TRANSCATÉTER CON TÉCNICA DE SUPERPOSICIÓN DE SENOS CORONARIOS FRENTE A LA TÉCNICA TRADICIONAL PARA REDUCCIÓN DE LA INCIDENCIA DEL BLOQUEO INTERAURICULAR RELACIONADA AL PROCEDIMIENTO: RESULTADOS DE UN ESTUDIO OBSERVACIONAL PROSPECTIVO EMPAREJADO POR PROPENSIÓN
- 5024-5 - REDUCCIÓN DE LA HOSPITALIZACIÓN CARDIOVASCULAR Y POR CUALQUIER CAUSA TRAS IMPLANTE DE VÁLVULA AÓRTICA TRANSCATÉTER MEDIANTE TÉCNICA DE SUPERPOSICIÓN DE SENOS CORONARIOS: RESULTADOS TRAS UN AÑO DE SEGUIMIENTO
-
Soroa Ortuño, Miguel
- 6055-2 - KCNH2 P. GLY262ALAFSTER98: UNA NUEVA VARIANTE ASOCIADA AL SÍNDROME DE QT LARGO EN UNA COHORTE ESPAÑOLA
- 5031-7 - GEMELOS EN LA MIOCARDIOPATÍA HIPERTRÓFICA. FENOTIPOS OPUESTOS PESE A GENÉTICA IDÉNTICA Y AMBIENTE SIMILAR
- 6044-10 - IMPLANTE DE VÁLVULA AÓRTICA TRANSCATÉTER CON TÉCNICA DE SUPERPOSICIÓN DE SENOS CORONARIOS FRENTE A LA TÉCNICA TRADICIONAL PARA REDUCCIÓN DE LA INCIDENCIA DEL BLOQUEO INTERAURICULAR RELACIONADA AL PROCEDIMIENTO: RESULTADOS DE UN ESTUDIO OBSERVACIONAL PROSPECTIVO EMPAREJADO POR PROPENSIÓN