Volumen 78. Número 5 (Mayo 2025)

Factor de impacto 2023
7,2
ISSN: 0300-8932
Artículos originales
- Los indicadores de calidad de la Sociedad Europea de Cardiología en fibrilación auricular en centros de excelencia en España: el registro SEC-EXCELENTE FA
- Martín Ruiz Ortiz, Elena Arbelo, Inmaculada Roldán Rabadán, Francisco Marín, Alejandro Pérez Cabeza, Raquel Marzoa Rivas, Rafael Peinado Peinado, Almudena Valle Alberca, Alicia Ibáñez Criado, Alfonso Valle Muñoz, Joaquín Osca Asensi, Ana del Río Lechuga, Francisco Javier Elola Somoza, Manuel Anguita Sánchez, en representación de los investigadores del registro SEC-EXCELENTE FA
- Rev Esp Cardiol. 2025;78:389-400
- Guías de práctica clínica e indicadores de calidad: ¿practicamos lo que predicamos?
- Finn Åkerström, Emma Svennberg
- Rev Esp Cardiol. 2025;78:401-3
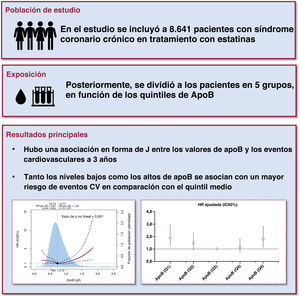
- Asociación en J entre la apolipoproteína B y resultados CV en pacientes con síndrome coronario crónico tratados con estatinas
- Jining He, Zhangyu Lin, Chenxi Song, Sheng Yuan, Xiaohui Bian, Bowen Li, Wenjun Ma, Kefei Dou
- Rev Esp Cardiol. 2025;78:404-13
- ApoB y pronóstico de los pacientes con síndrome coronario crónico
- Alberto Cordero, Rosa Fernández Olmo
- Rev Esp Cardiol. 2025;78:414-5
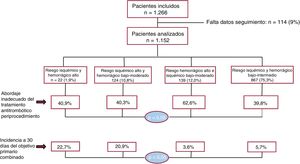
- Adherencia a las recomendaciones de tratamiento antitrombótico periprocedimiento e impacto pronóstico en pacientes con alto riesgo isquémico y hemorrágico
- María Anguita-Gámez, David Vivas, Raquel Ferrandis, María Asunción Esteve-Pastor, Rafael González-Manzanares, Marysol Echeverri, Jesús Igualada, Isabel Egocheaga, Beatriz Nozal-Mateo, Ane Abad-Motos, Elena Figuero, Nuria Bouzó-Molina, Teresa Lozano, Carlos Álvarez-Ortega, Javier Torres, María José Descalzo, Juan Carlos Catalá, Enrique Martín-Rioboo, Alejandra Moliner, Rocío Rodríguez-Contreras, Manuel Carnero-Alcázar, Francisco Marín, Manuel Anguita, en representación de los investigadores del estudio REQXAA
- Rev Esp Cardiol. 2025;78:416-24
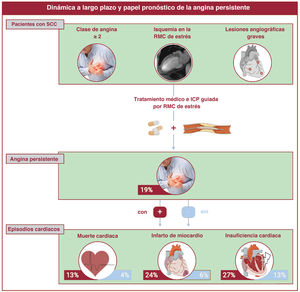
- Utilidad pronóstica de la angina persistente tras revascularización percutánea en el síndrome coronario crónico con angiografía y RMC de estrés alteradas
- Nerea Pérez-Solé, Elena de Dios, José V. Monmeneu, María P. López-Lereu, José Gavara, César Ríos-Navarro, Víctor Marcos-Garces, Héctor Merenciano, Clara Bonanad, Joaquim Cánoves, Félix Platero, Andrea Ventura, David Moratal, Antoni Bayés-Genís, Jorge Sanz, Manuel Jiménez-Navarro, Luis Martínez-Dolz, Juan Sanchis, Julio Núñez, Vicente Bodí
- Rev Esp Cardiol. 2025;78:425-36
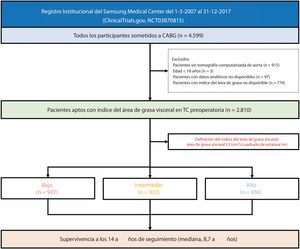
- Impacto clínico de la adiposidad visceral en la mortalidad a largo plazo de pacientes sometidos a revascularización coronaria
- Jinhwan Jo, Seung Hun Lee, Jeong Hoon Yang, Sung Mok Kim, Ki Hong Choi, Young Bin Song, Dong Seop Jeong, Joo Myung Lee, Taek Kyu Park, Joo-Yong Hahn, Seung-Hyuk Choi, Su Ryeun Chung, Yang Hyun Cho, Kiick Sung, Wook Sung Kim, Hyeon-Cheol Gwon, Young Tak Lee
- Rev Esp Cardiol. 2025;78:437-46
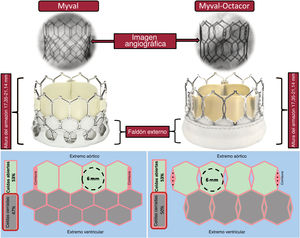
- Nueva prótesis percutánea expandible con balón Myval: revisión sistemática de las indicaciones aórtica, mitral, tricuspídea y pulmonar
- Mario García-Gómez, Clara Fernández-Cordón, José Carlos González-Gutiérrez, Ana Serrador, Alberto Campo, Carlos Cortés Villar, Sara Blasco Turrión, Cristhian Aristizábal, Julio Peral Oliveira, Alexander Stepanenko, Mikel González Arribas, Luca Scorpiglione, Akash Jain, David Carnicero Martínez, J. Alberto San Román, Ignacio J. Amat-Santos
- Rev Esp Cardiol. 2025;78:447-64
Arritmias y estimulación cardiaca

Comentario editorial
Cardiopatía isquémica
Comentario editorial
Epidemiología, factores de riesgo y prevención
Cardiología intervencionista
Artículos de revisión
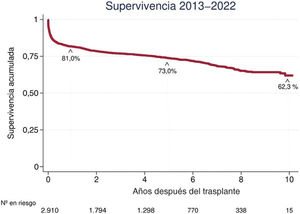
- Trasplante cardiaco como tratamiento del shock cardiogénico: ¿una estrategia eficiente o una «excepción española»?
- Francisco José Hernández-Pérez, Alba Martín-Centellas, Mercedes Rivas-Lasarte, Cristina Mitroi, Manuel Gómez-Bueno, Javier Segovia-Cubero
- Rev Esp Cardiol. 2025;78:465-72
Articles in press
- Afectación cardiaca poco común en osteosarcoma metastásico
- Juan Manuel Serrano-Marcos, Víctor Manuel López-Espinosa, Míriam Cantero-Campos
- 10.1016/j.recesp.2025.04.008
- Pruebas preliminares. Disponible online: 12 mayo 2025
- Atresia pulmonar con septo interventricular intacto
- Alejandro Fernández-Cisneros, Bosco A. Moscoso, Stefano Congiu
- 10.1016/j.recesp.2025.02.023
- Pruebas preliminares. Disponible online: 12 mayo 2025
- Del estetoscopio a la inteligencia artificial: decodificando la enfermedad tricuspídea
- Covadonga Fernández-Golfín, Ana García-Martín
- 10.1016/j.recesp.2025.03.012
- Disponible online: 12 mayo 2025
- Validación de la secuencia de resonancia magnética 3D de única apnea ESSOS para el estudio de la dilatación aórtica
- Sandra Gómez-Talavera, Álvaro Navarro-Guzmán, Rodrigo Fernández-Jiménez, Valentín Fuster, Javier Sánchez-González, Borja Ibáñez
- 10.1016/j.recesp.2025.04.009
- Pruebas preliminares. Disponible online: 9 mayo 2025

Revista Española de Cardiología 75 años en el corazón de la investigación cardiovascular
El coloquio « Revista Española de Cardiología : 75 años en el corazón de la investigación cardiovascular» rememora los logros, dificultades y desafíos de su recorrido único a través de los editores jefe desde los años 90.
Juan Sanchis
Editor jefe de Revista Española de Cardiología (2015-2021-)
Francisco Fernández-Avilés
Editor jefe de Revista Española de Cardiología (1991-1997)
Xavier Bosch
Editor jefe de Revista Española de Cardiologíaz (1997-2003)
Fernando Alfonso
Editor jefe de Revista Española de Cardiología (2004-2009)
Ignacio Ferreira
Editor jefe de Revista Española de Cardiología (2015-2021)