Volume 77. Number 10 (October 2024)

Impact factor 2023
7.2
ISSN: 1885-5857

Original articles
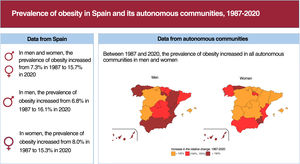
- Prevalence of obesity in Spain and its autonomous communities, 1987-2020
- Laura Feijoo, Julia Rey-Brandariz, Carla Guerra-Tort, Cristina Candal-Pedreira, María Isolina Santiago-Pérez, Alberto Ruano-Ravina, Mónica Pérez-Ríos
- Rev Esp Cardiol. 2024;77:809-18
- Obesity in Spain: an open book that must be read
- Emilio Ortega Martínez de Victoria, Adriana Pané Vila, Amanda Jiménez Pineda
- Rev Esp Cardiol. 2024;77:819-20
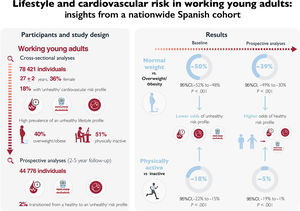
- Lifestyle and cardiovascular risk in working young adults: insights from a nationwide Spanish cohort
- Adrián Castillo-García, Pedro L. Valenzuela, Gonzalo Saco-Ledo, Pedro Carrera-Bastos, Luis M. Ruilope, Alejandro Santos-Lozano, Alejandro Lucia
- Rev Esp Cardiol. 2024;77:821-31
- Unhealthy lifestyle and cardiovascular risk profile: a concern in working young adults in Spain
- Dimelza Osorio-Sánchez
- Rev Esp Cardiol. 2024;77:832-4
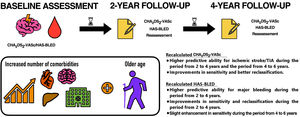
- Dynamic assessment of CHA2DS2-VASc and HAS-BLED scores for predicting ischemic stroke and major bleeding in atrial fibrillation patients
- María José Serna, José Miguel Rivera-Caravaca, Raquel López-Gálvez, Eva Soler-Espejo, Gregory Y.H. Lip, Francisco Marín, Vanessa Roldán
- Rev Esp Cardiol. 2024;77:835-42
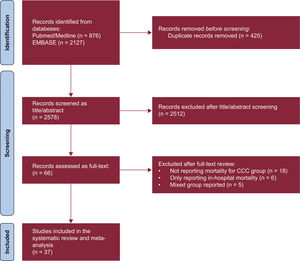
- A systematic review and meta-analysis of mortality in chronic Chagas cardiomyopathy versus other cardiomyopathies: higher risk or fiction?
- Sergio A. Gómez-Ochoa, Angie Yarlady Serrano-García, Alexandra Hurtado-Ortiz, Andrea Aceros, Lyda Z. Rojas, Luis E. Echeverría
- Rev Esp Cardiol. 2024;77:843-50
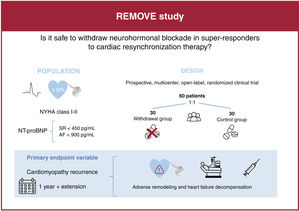
- Withdrawal of drug therapy in responders to cardiac resynchronization therapy: rationale and design of the REMOVE trial
- Francisco J. Pastor-Pérez, Iris P. Garrido-Bravo, Pablo Peñafiel-Verdú, Noelia Fernández-Villa, Sergio Manzano-Fernández, María José Oliva-Sandoval, María Teresa Pérez-Martínez, César Caro-Martínez, Álvaro Hernández-Vicente, Domingo A. Pascual-Figal, on behalf of the clinical trial REMOVE researches
- Rev Esp Cardiol. 2024;77:851-8
- Valvulitis: a new echocardiographic criterion for the diagnosis of bioprosthetic aortic valve infective endocarditis
- Pablo Zulet, Isidre Vilacosta, Eduardo Pozo, Daniel García-Arribas, Carlos Nicolás Pérez-García, Manuel Carnero, Daniel Pérez-Camargo, Lourdes Montero, Melchor Saiz-Pardo, Patricia Mahía, Adrián Jerónimo, Fabián Islas, Daniel Gómez, José Alberto San Román, José Alberto de Agustín, Carmen Olmos
- Rev Esp Cardiol. 2024;77:859-67
Epidemiology, risk factors and prevention
Editorial comment
Editorial comment
Arrythmias and cardiac stimulation
Cardiomyopathy and heart failure
Valvular Heart Disease
Editorials
- Ethics in clinical research: what you should know before starting a study
- Armando Pérez de Prado, Almudena Castro Conde, Ignacio Ferreira-González
- Rev Esp Cardiol. 2024;77:805-8
Articles in press
- Lumenless versus stylet-driven leads in left bundle branch pacing
- Álvaro Marco Del Castillo, Javier Ramos Jiménez, Luis Borrego Bernabé, Fernando Arribas Ynsaurriaga, Daniel Rodríguez Muñoz, Rafael Salguero Bodes
- 10.1016/j.rec.2024.08.014
- Journal pre-proofs. Available online: 23 October 2024
- Management of heart disease in renal transplant recipients: a national Delphi survey-based SET/SEC/SEN consensus document
- María Dolores García-Cosío, Josep María Cruzado, Marta Farrero, María Teresa Blasco Peiró, Marta Crespo, Juan Francisco Delgado Jiménez, Beatriz Díaz Molina, Constantino Fernández Rivera, Iris Paula Garrido Bravo, Verónica López Jiménez, Edoardo Melilli, Sonia Mirabet Pérez, María Lourdes Pérez Tamajón, Diego Rangel Sousa, Emilio Rodrigo Calabia, Domingo Hernández Marrero
- 10.1016/j.rec.2024.09.008
- Journal pre-proofs. Available online: 23 October 2024
- ApoB and prognosis of patients with chronic coronary syndrome
- Alberto Cordero, Rosa Fernández Olmo
- 10.1016/j.rec.2024.10.003
- Journal pre-proofs. Available online: 22 October 2024
- Coronary plaque modification and impact on the microcirculation territory after drug-coated balloon angioplasty: the PLAMI study
- José Antonio Sorolla Romero, Andrea Teira Calderón, Jean Paul Vilchez Tschischke, José Luis Díez Gil, Hector M. Garcia-Garcia, Jorge Sanz Sánchez
- 10.1016/j.rec.2024.10.001
- Journal pre-proofs. Available online: 22 October 2024