Volumen 78. Número 7 (Julio 2025)

Factor de impacto 2024
4,9
ISSN: 0300-8932
Artículos originales
-
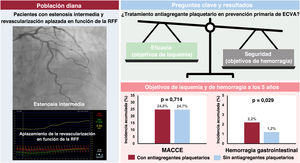
- Seguridad y eficacia del tratamiento antiplaquetario en pacientes con estenosis coronaria intermedia y revascularización diferida
- David Hong, Seung Hun Lee, Jihye Heo, Doosup Shin, Juhee Cho, Eliseo Guallar, Hyun Sung Joh, Hyun Kuk Kim, Junho Ha, Ki Hong Choi, Taek Kyu Park, Jeong Hoon Yang, Young Bin Song, Joo-Yong Hahn, Seung-Hyuk Choi, Hyeon-Cheol Gwon, Danbee Kang, Joo Myung Lee
- Rev Esp Cardiol. 2025;78:580-9
Comentario editorial
- Tratamiento antiagregante en la prevención primaria de lesiones coronarias no significativas
- Jose Antonio Esteban-Chapel, Juan Antonio Franco-Peláez, Alvaro Aceña
- Rev Esp Cardiol. 2025;78:590-1
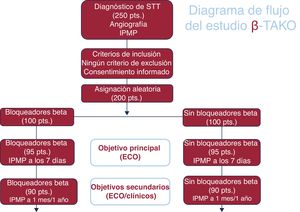
- Justificación y diseño del estudio sobre bloqueadores beta en el síndrome de tako-tsubo: un estudio clínico aleatorizado (β-Tako)
- Fernando Alfonso, Jorge Salamanca, Iván Núñez-Gil, Borja Ibáñez, Juan Sanchis, Manel Sabaté, Maite Velázquez, Sergio Raposeiras-Roubín, Tamara García-Camarero, Paula Antuña, Hernán Mejía, Xavier Carrillo, Irene Buera, Manuel Martínez-Sellés, Juan Manuel Escudier-Villa, Joaquín Sánchez-Prieto, Emilia Blanco Ponce, Gonzalo Cabezón, Covadonga Fernández-Golfín, Domingo Pascual-Figal, Belén Cid, Ana Marcano, Rafael González-Manzanares, Santiago Jiménez-Valero, José Manuel Vázquez, Jorge Sanz-Sánchez, Alberto Cecconi, David Del Val, Francisco Abad-Santos, Filippo Crea
- Rev Esp Cardiol. 2025;78:592-9
-

- Calidad del sueño e hipertensión incidental
- Zhihao Zheng, Yanjun Song, Zechen Liu, Jining He, Shanshan Shi, Chenxi Song, Rui Fu, Lei Jia, Guofeng Gao, Qiuting Dong, Min Yang, Wenjun Ma, Kefei Dou
- Rev Esp Cardiol. 2025;78:600-8
-
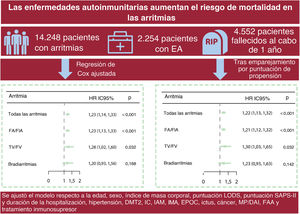
- Asociación entre enfermedades autoinmunitarias y mortalidad por cualquier causa en pacientes con arritmias cardiacas
- Le Li, Lingmin Wu, Zhicheng Hu, Limin Liu, Likun Zhou, Zhuxin Zhang, Minghao Zhao, Yulong Xiong, Zhenhao Zhang, Lihui Zheng, Ligang Ding, Yan Yao
- Rev Esp Cardiol. 2025;78:609-17
-
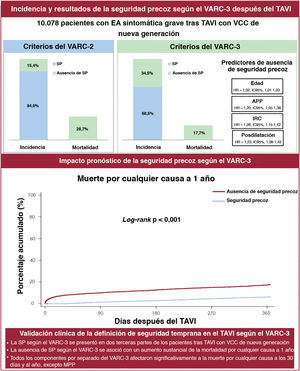
- Seguridad precoz tras TAVI según los criterios VARC-3: incidencia, predictores e impacto clínico
- Ariana Gonzálvez-García, Pedro Cepas-Guillén, Julien Ternacle, Marina Urena, Alberto Alperi, Asim N. Cheema, Gabriela Veiga-Fernández, Luis Nombela-Franco, Victoria Vilalta, Giovanni Esposito, Francisco Campelo-Parada, Ciro Idolfi, María del Trigo, Antonio Muñoz-García, Nicolás Maneiro, Luis Asmarats, Ander Regueiro, David del Val, Vicenç Serra, Vincent Auffret, Melchior Jonveaux, Guillaume Bonnet, Jules Mesnier, Suc Gaspard, Pablo Avanzas, Effat Rezaei, Víctor Fradejas-Sastre, Gabriela Tirado-Conte, Eduard Fernández-Nofrerías, Anna Franzone, Thibaut Guitteny, Sabato Sorrentino, Juan Francisco Oteo, Felipe Díez-Delhoyo, Lola Gutiérrez-Alonso, Pablo Vidal, Fernando Alfonso, Andrea Monastyrski, Maxime Nolf, Emilie Pelletier-Beaumont, Marisa Avvedimento, Josep Rodés-Cabau
- Rev Esp Cardiol. 2025;78:618-27
Cardiopatía isquémica y cuidados agudos cardiovasculares
Epidemiología, factores de riesgo y prevención
Arritmias y estimulación cardiaca
Cardiología intervencionista
Artículos especiales
-
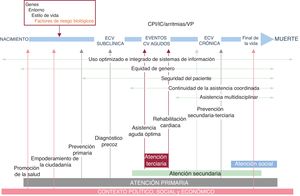
- Desarrollo y puesta en marcha de un plan nacional de salud cardiovascular. Estrategia en salud cardiovascular (ESCAV) española
- Héctor Bueno, Germán Seara, María Rosario Azcutia, María Jesús Rodríguez-García, Sonia Peláez, Yolanda Agra, Carla A. Dueñas, Pedro Gullón, Pilar Aparicio Azcárraga, en representación del Comité Asesor Multidisciplinar de la Estrategia en Salud Cardiovascular del Sistema Nacional de Salud (ESCAV)
- Rev Esp Cardiol. 2025;78:628-36
Comentario editorial
- La Estrategia en Salud Cardiovascular del Sistema Nacional de Salud (ESCAV). Una gran oportunidad y responsabilidad para todos
- Luis Rodríguez Padial, Julián Pérez Villacastín
- Rev Esp Cardiol. 2025;78:637-8
-
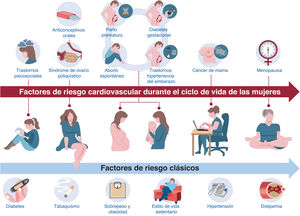
- Prevención cardiovascular primaria y secundaria en los ciclos vitales de la mujer. Documento de consenso de la SEC-GT ECV en la Mujer, ACP-SEC, SEGO, AEEM, SEEN, semFYC, SEMERGEN, AEP y AEM
- Antonia Sambola, Raquel Campuzano, Almudena Castro, María Goya, Pluvio Coronado, Rosa Fernández-Olmo, Miguel Ángel María-Tablado, Carolina Ortiz-Cortés, Xènia Ortolà, Vicente Pallarés-Carratalá, Antonia Pijuan-Domenech, Rosa M. Plata, Rosa María Sánchez-Hernández, José Manuel Siurana, Càtia Timoteo, Begoña Viejo-Hernández
- Rev Esp Cardiol. 2025;78:639-51
Articles in press
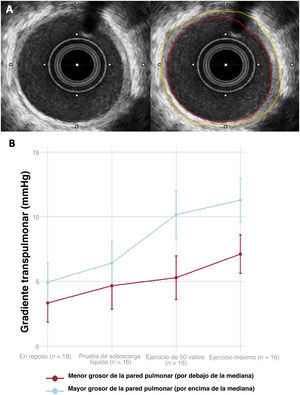
- Evaluación del remodelado vascular pulmonar adverso mediante ecografía intravascular en la circulación de Fontan
- Yassin Belahnech, Gerard Martí-Aguasca, Enric Domingo-Ribas, Eduard Ródenas-Alesina, Antonia Pijuan-Domenech, Laura Dos-Subirà
- 10.1016/j.recesp.2025.05.004
- Disponible online: 8 julio 2025
- Puente epicárdico de aleteo auricular. Al alcance del catéter
- Alejandro Carta-Bergaz, Ángel Arenal-Maíz
- 10.1016/j.recesp.2025.04.007
- Disponible online: 8 julio 2025
- El diagnóstico etiológico en miocarditis: ¿una asignatura aún pendiente?
- Fernando Domínguez
- 10.1016/j.recesp.2025.06.011
- Pruebas preliminares. Disponible online: 7 julio 2025
- Estrategia de reducción de stents frente a revascularización coronaria percutánea convencional en el IAMCEST: el ensayo COPERNICAN
- Jorge Sanz-Sánchez, Sandra Santos Martínez, Eva Rumiz González, Juan Francisco Oteo Domínguez, David Tejada Ponce, Antonio Gómez Menchero, Guillermo Sánchez Elvira, Georgina Fuertes Ferre, Fernando Rivero Crespo, Antonela Lukic Otanovic, José Díaz Fernández, Eladio Galindo Fernández, Cristóbal Urbano Carrillo, Neus Salvatella Giralt, Mauricio Torres Sánchez, Arturo García Touchard, Borja Ibáñez Cabeza, Giulio Stefanini, Fernando Alfonso Manterola, Héctor García García, Ignacio J. Amat-Santos
- 10.1016/j.recesp.2025.05.006
- Pruebas preliminares. Disponible online: 7 julio 2025

Revista Española de Cardiología 75 años en el corazón de la investigación cardiovascular
El coloquio « Revista Española de Cardiología : 75 años en el corazón de la investigación cardiovascular» rememora los logros, dificultades y desafíos de su recorrido único a través de los editores jefe desde los años 90.
Juan Sanchis
Editor jefe de Revista Española de Cardiología (2015-2021-)
Francisco Fernández-Avilés
Editor jefe de Revista Española de Cardiología (1991-1997)
Xavier Bosch
Editor jefe de Revista Española de Cardiologíaz (1997-2003)
Fernando Alfonso
Editor jefe de Revista Española de Cardiología (2004-2009)
Ignacio Ferreira
Editor jefe de Revista Española de Cardiología (2015-2021)