ISSN: 0300-8932
Factor de impacto 2024
4,9
SEC 2019 - El Congreso de las Enfermedades Cardiovasculares

Barcelona,
17 - 19 de Octubre de 2019
Introducción
Dr. Arturo Evangelista Masip
Presidente del Comité Científico del Congreso
Comité ejecutivo
Comité de evaluadores
Índice de autores
5005. It runs in the family
Fecha
: 17-10-2019 10:45:00
Tipo
: Comunicaciones mini orales
Sala
: Sala 8 (Nivel 2)
5005-2. ¿CUÁNDO SOLICITAR UN ESTUDIO GENÉTICO EN LA MIOCARDIOPATÍA DILATADA? PREDICTORES DE UN RESULTADO GENÉTICO POSITIVO
Marta López Serna, María Gallego Delgado, Eduardo Villacorta Argüelles, Belén García Berrocal, Elena Díaz Peláez, Marta Alonso Fernández de Gatta, Lucía Rodríguez Estévez, Elena Marcos Vadillo, Ana Martín García, Elisabete Alzola Martínez de Antoñana, Alfredo Barrio Rodríguez, María Isidoro García y Pedro Luis Sánchez Fernández, del Complejo Asistencial Universitario de Salamanca, Salamanca.
Introducción y objetivos: La miocardiopatía dilatada (MCD) puede tener un origen familiar. Las herramientas diagnósticas para su estudio son la historia clínica individual y familiar, el estudio de familiares y el análisis genético. El objetivo de este trabajo fue identificar predictores de un resultado genético positivo en pacientes con MCD.
Métodos: Analizamos los datos demográficos, clínicos y pruebas complementarias habituales de todos los casos índices con MCD referidos a una consulta de cardiopatías familiares a los que se realizó estudio genético.
Resultados: Se estudiaron 101 casos índice de MCD (66% varones, edad media diagnóstico 56 ± 15 años; 80% con resonancia magnética cardiaca (RMC)). Tras la evaluación 47 (46%) fueron MCD familiares, 13 (13%) fueron MCD atribuibles a otras causas y 41 (41%) permanecieron sin etiología identificable. El estudio genético fue positivo en el 24% (51% de MCD familiares), siendo el gen con mutaciones patogénicas más frecuente TTN (figura). En la tabla se muestra un resumen del análisis univariante. Los pacientes con resultados genéticos positivos tenían más antecedentes familiares de muerte súbita (p = 0,023), de implante de marcapasos precoz (< 60 años) (p < 0,001), o trasplante cardiaco (p < 0,001), y mayor presencia de TVNS/EV alta densidad en el Holter (p = 0,007). En relación con la RMC, tenían menor volumen telediastólico indexado, geometría de VI normal y mayor fracción de volumen extracelular. El antecedente familiar de muerte súbita (definido como muerte súbita en familiar de primer grado a edad < 40 años o con MCD) fue el único predictor independiente de un resultado genético positivo (OR = 6,3; p < 0,05).
|
Análisis univariante para resultado genético |
|||||||
|
G+ |
G no + |
p |
G+ |
G no + |
p |
||
|
Edad, años |
55 ± 15 |
56 ± 15 |
NS |
CK |
116 (62-192) |
110 (65-163) |
NS |
|
Varón |
15 (63%) |
52 (68%) |
NS |
VTDVI indexado, ml/m2 |
111 ± 31 |
141 ± 46 |
0,014 |
|
AF MS |
6 (25%) |
6 (8%) |
0,023 |
FEVI, % |
34 ± 11 |
32 ± 11 |
NS |
|
AF TxC |
6 (25%) |
0 (0%) |
0,000 |
Disfunción biventricular |
6 (35%) |
19 (34%) |
NS |
|
AF MCp <60 años |
4 (17%) |
0 (0%) |
0,000 |
Hipertrofia VI excéntrica |
7 (41%) |
37 (70%) |
0,033 |
|
BRIHH |
11 (46%) |
46 (60%) |
NS |
No compactación |
2 (12%) |
6 (10,5%) |
NS |
|
BAV |
3 (12,5%) |
10 (13%) |
NS |
Realce tardío |
8 (50%) |
26 (45%) |
NS |
|
TVS/FV |
4 (17%) |
8 (10%) |
NS |
VEC, % |
0,317 ± 0,71 |
0,272 ± 0,04 |
0,024 |
|
TVNS/EV |
9 (37,5%) |
10 (13%) |
0,007 |
T1 nativo, ms |
1059 ± 81 |
1050 ± 33 |
NS |
|
VEC: volumen extracelular; G+: genética positiva; NS: no significativo; TxC: trasplante cardiaco; VTDVI: volumen telediastólica ventricular izquierdo. |
|||||||
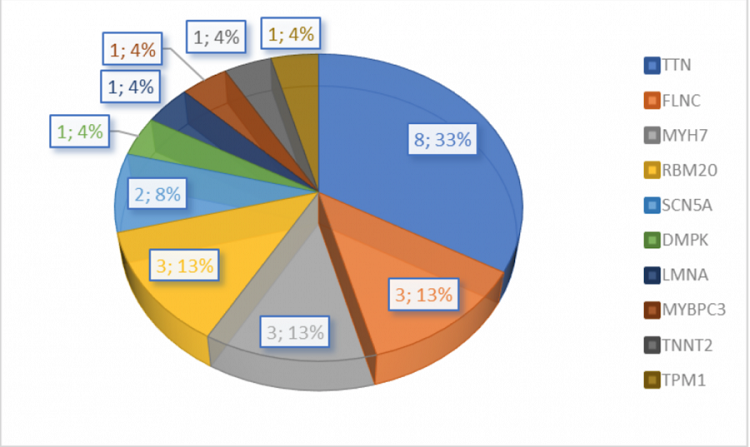
Distribución de genes con mutaciones patogénicas.
Conclusiones: La historia familiar y el estudio clínico de los familiares, junto con la genética son las herramientas más útiles para identificar pacientes con una MCD hereditaria, siendo el antecedente familiar de muerte súbita el predictor independiente de un resultado genético positivo. En nuestra muestra, casi en la mitad de las MCD familiares no se identificó una mutación causal.
Comunicaciones disponibles de "<i>It runs in the family</i>"
- 5005-1. MODERADORES
- Esther González López, Madrid, y Julián Palomino Doza, Valladolid.
- 5005-2. ¿CUÁNDO SOLICITAR UN ESTUDIO GENÉTICO EN LA MIOCARDIOPATÍA DILATADA? PREDICTORES DE UN RESULTADO GENÉTICO POSITIVO
- Marta López Serna, María Gallego Delgado, Eduardo Villacorta Argüelles, Belén García Berrocal, Elena Díaz Peláez, Marta Alonso Fernández de Gatta, Lucía Rodríguez Estévez, Elena Marcos Vadillo, Ana Martín García, Elisabete Alzola Martínez de Antoñana, Alfredo Barrio Rodríguez, María Isidoro García y Pedro Luis Sánchez Fernández, del Complejo Asistencial Universitario de Salamanca, Salamanca.
- 5005-3. VARIANTES GENÉTICAS CON IMPACTO PRONÓSTICO EN MIOCARDIOPATÍA HIPERTRÓFICA. HISTORIA NATURAL DE 4 FAMILIAS CON MUTACIONES EN LA ARGININA 719 DE MYH7
- María Tamargo Delpón, M. Ángeles Espinosa Castro, Irene Méndez Fernández, Sofía Cuenca Parra, Rebeca Lorca Gutiérrez, Ana Isabel Fernández, Nélida Vázquez Aguilera, Raquel Yotti Álvarez, Javier Bermejo Thomas y Francisco Fernández Avilés, del Hospital General Universitario Gregorio Marañón, Madrid.
- 5005-4. PREDICTORES CLÍNICOS DE MIOCARDIOPATÍA GENÉTICA DE ALTO RIESGO ARRÍTMICO EN PACIENTES CON MIOCARDIOPATÍA DILATADA NO ISQUÉMICA
- Eva Cabrera Borrego, Francisco José Bermúdez-Jiménez, Lorena González Camacho, Rosa Macías Ruíz, Diego Segura Rodríguez, Javier Ramos Maqueda, Mercedes Cabrera Ramos, Miguel Álvarez López, Pablo J. Sánchez Millán, Laura Jordán Martínez, Miguel Morales García, Rocío Parrilla Linares, Ricardo Rivera López, Manuel Molina Lerma y Juan Jiménez Jáimez, del Hospital Universitario Virgen de las Nieves, Granada.
- 5005-5. VARIANTES GENÉTICAS ASOCIADAS CON LA MIOCARDIOPATÍA INDUCIDA POR QUIMIOTERAPIA
- María Alejandra Restrepo Córdoba1, Yuri Kim2, Beatriz Núñez-García1, Fernando Domínguez3, Antoni Bayés-Genís4, Alfredo Bardají5, Domingo Pascual-Figal6, José Manuel García-Pinilla7, Isabel Serrano5, Josep Lupón Rosés4, Mariano Provencio1, Richard Aplenc8, James Ware9, Christine E Seidman2 y Pablo García-Pavía3, del 1Hospital Universitario Puerta de Hierro, Majadahonda (Madrid), 2Harvard Medical School, Boston Massachusetts (EE.UU.), 3Hospital Universitario Puerta de Hierro, Majadahonda (Madrid), 4Hospital Universitari Germans Trias i Pujol, Badalona (Barcelona), 5Hospital Universitario Joan XXIII, Tarragona, 6Hospital Clínico Universitario Virgen de la Arrixaca, El Palmar (Murcia), 7Hospital Clínico Universitario Virgen de la Victoria, Málaga, 8Perelman School of Medicine and University of Pennsylvania Health System, Philadelphia, Pennsylvania (EE.UU.) y 9Royal Brompton and Harefield NHS Foundation Trust, London (Reino Unido).
- 5005-6. NUEVA MUTACIÓN EN EL GEN HCN4 EN UNA FAMILIA CON BRADICARDIA SINUSAL COMBINADA CON NO COMPACTACIÓN DEL VENTRÍCULO IZQUIERDO
- Eduardo Villacorta Argüelles1, Ricardo Caballero2, Teresa Crespo-García2, María Gallego Delgado1, Marta Alonso Fernández de Gatta1, Belén García Berrocal3, Elena Marcos Vadillo3, Belén Plata Izquierdo4, María Isidoro García3, Juan Tamargo Menéndez2, Eva Delpón Mosquera2 y Pedro Luis Sánchez Fernández1, del 1Servicio de Cardiología, Complejo Asistencial Universitario de Salamanca, Salamanca, 2Grupo de Investigación Farmacología Cardiovascular, Universidad Complutense de Madrid, Madrid, 3Servicio de Bioquímica Clínica, Complejo Asistencial Universitario de Salamanca, Salamanca, 4Servicio de Pediatría, Complejo Asistencial Universitario de Salamanca, Salamanca.
- 5005-7. ESTUDIO FAMILIAR Y GENÉTICO EN LAS CARDIOPATÍAS HEREDITARIAS
- Elisa Nicolás Rocamora1, María Orenes1, Cristina Gil1, María del Carmen Olmo Conesa1, David López Cuenca1, Carmen Muñoz Esparza1, Marina Navarro1, Juan José Santos Mateo1, Juan Ramón Gimeno Blanes1 y María Sabater Molina2, del 1Hospital Clínico Universitario Virgen de la Arrixaca, El Palmar (Murcia) y 2Instituto Murciano de investigación Biosanitaria IMIB-Arrixaca, Murcia.
- 5005-8. CRIBADO GENÉTICO DE LOS PRINCIPALES GENES ASOCIADOS A PATOLOGÍAS CARDIOVASCULARES MEDIANTE SECUENCIACIÓN MASIVA EN UNA COHORTE DE PACIENTES TRASPLANTADOS DE CORAZÓN
- Elías Cuesta Llavona1, Juan Gómez de Oña1, Beatriz Díaz Molina1, José Luis Lambert Rodríguez1, M. José Bernardo Rodríguez1, Belén Alonso González1, Sara Iglesias Álvarez1, Rebeca Lorca Gutiérrez2, José Julián Rodríguez Reguero1 y Eliecer Coto García1, del 1Hospital Universitario Central de Asturias, Oviedo (Asturias) y 2Hospital General Universitario Gregorio Marañón, Madrid.
Más comunicaciones de los autores
-
Alonso Fernández de Gatta, Marta
- 6032-286 - TOMOGRAFÍA COMPUTARIZADA CARDIOVASCULAR GESTIONADA POR CARDIOLOGÍA Y SU PAPEL DENTRO DE UN PROTOCOLO DE UNIDAD DE DOLOR TORÁCICO
- 4031-6 - SÍNDROME MIELODISPLÁSICO BAJO SOPORTE TRANSFUSIONAL: NUEVA ESCALA PREDICTIVA DE MUERTE Y EVENTOS CARDIOVASCULARES
- 6010-99 - APIXABÁN FRENTE A ACENOCUMAROL EN LA CARDIOVERSIÓN ELÉCTRICA DE PACIENTES CON FIBRILACIÓN AURICULAR: DEL EMANATE A LA VIDA REAL
- 4001-9 - FACTORES RELACIONADOS CON EL DESTETE EXITOSO DE OXIGENADOR CON MEMBRANA EXTRACORPÓREA VENOARTERIAL. EXPERIENCIA EN UN HOSPITAL TERCIARIO
- 4003-7 - EVALUACIÓN DE PARÁMETROS ECOCARDIOGRÁFICOS PREDICTORES EN EL DESTETE DE OXIGENADOR EXTRACORPÓREO DE MEMBRANA VENOARTERIAL
- 4031-4 - ANÁLISIS DE LOS BIOMARCADORES CARDIACOS EN EL DIAGNÓSTICO DE SIDEROSIS CARDIACA Y SU VALOR PRONÓSTICO EN EL SÍNDROME MIELODISPLÁSICO DE BAJO GRADO RIESGO
- 5005-2 - ¿CUÁNDO SOLICITAR UN ESTUDIO GENÉTICO EN LA MIOCARDIOPATÍA DILATADA? PREDICTORES DE UN RESULTADO GENÉTICO POSITIVO
- 5005-6 - NUEVA MUTACIÓN EN EL GEN HCN4 EN UNA FAMILIA CON BRADICARDIA SINUSAL COMBINADA CON NO COMPACTACIÓN DEL VENTRÍCULO IZQUIERDO
- 5007-7 - INTELIGENCIA ARTIFICIAL FRENTE A ESTADÍSTICA CLÁSICA EN LA PREDICCIÓN DE CARDIOVERSIÓN ESPONTÁNEA, FARMACOLÓGICA, ELÉCTRICA Y RECURRENCIA DE FIBRILACIÓN AURICULAR
- 7002-7 - MINOCA: CARACTERIZACIÓN CLÍNICA Y PAPEL DE LA RESONANCIA MAGNÉTICA CARDIACA EN EL DIAGNÓSTICO ETIOLÓGICO DEFINITIVO
-
Alzola Martínez de Antoñana, Elisabete
- 5005-2 - ¿CUÁNDO SOLICITAR UN ESTUDIO GENÉTICO EN LA MIOCARDIOPATÍA DILATADA? PREDICTORES DE UN RESULTADO GENÉTICO POSITIVO
- 4001-9 - FACTORES RELACIONADOS CON EL DESTETE EXITOSO DE OXIGENADOR CON MEMBRANA EXTRACORPÓREA VENOARTERIAL. EXPERIENCIA EN UN HOSPITAL TERCIARIO
- 4003-7 - EVALUACIÓN DE PARÁMETROS ECOCARDIOGRÁFICOS PREDICTORES EN EL DESTETE DE OXIGENADOR EXTRACORPÓREO DE MEMBRANA VENOARTERIAL
- 6046-393 - ABLACIÓN DE SUSTRATO EN TORMENTA ARRÍTMICA BAJO SOPORTE CON ASISTENCIA CIRCULATORIA TIPO OXIGENADOR EXTRACORPÓREO DE MEMBRANA
-
Barrio Rodríguez, Alfredo
- 6046-393 - ABLACIÓN DE SUSTRATO EN TORMENTA ARRÍTMICA BAJO SOPORTE CON ASISTENCIA CIRCULATORIA TIPO OXIGENADOR EXTRACORPÓREO DE MEMBRANA
- 4001-9 - FACTORES RELACIONADOS CON EL DESTETE EXITOSO DE OXIGENADOR CON MEMBRANA EXTRACORPÓREA VENOARTERIAL. EXPERIENCIA EN UN HOSPITAL TERCIARIO
- 4003-7 - EVALUACIÓN DE PARÁMETROS ECOCARDIOGRÁFICOS PREDICTORES EN EL DESTETE DE OXIGENADOR EXTRACORPÓREO DE MEMBRANA VENOARTERIAL
- 5005-2 - ¿CUÁNDO SOLICITAR UN ESTUDIO GENÉTICO EN LA MIOCARDIOPATÍA DILATADA? PREDICTORES DE UN RESULTADO GENÉTICO POSITIVO
-
Díaz Peláez, Elena
- 6032-286 - TOMOGRAFÍA COMPUTARIZADA CARDIOVASCULAR GESTIONADA POR CARDIOLOGÍA Y SU PAPEL DENTRO DE UN PROTOCOLO DE UNIDAD DE DOLOR TORÁCICO
- 5019-5 - ANÁLISIS DE LA DEFORMACIÓN MIOCÁRDICA COMO FACTOR PREDICTOR PRONÓSTICO DE EVENTOS CARDIOVASCULARES EN EL TRASPLANTE DE PROGENITORES HEMATOPOYÉTICOS
- 6009-76 - EVALUACIÓN ECOCARDIOGRÁFICA BASAL EN ENFERMOS HEMATOLÓGICOS CANDIDATOS A TRASPLANTE ALOGÉNICO DE PRECURSORES HEMATOPOYÉTICOS
- 5019-8 - REMODELADO VENTRICULAR TRAS EL USO DE SACUBITRILO/VALSARTÁN EN LA MIOCARDIOPATÍA TÓXICA EN EL PACIENTE CON CÁNCER
- 5005-2 - ¿CUÁNDO SOLICITAR UN ESTUDIO GENÉTICO EN LA MIOCARDIOPATÍA DILATADA? PREDICTORES DE UN RESULTADO GENÉTICO POSITIVO
- 4031-6 - SÍNDROME MIELODISPLÁSICO BAJO SOPORTE TRANSFUSIONAL: NUEVA ESCALA PREDICTIVA DE MUERTE Y EVENTOS CARDIOVASCULARES
- 4031-4 - ANÁLISIS DE LOS BIOMARCADORES CARDIACOS EN EL DIAGNÓSTICO DE SIDEROSIS CARDIACA Y SU VALOR PRONÓSTICO EN EL SÍNDROME MIELODISPLÁSICO DE BAJO GRADO RIESGO
-
Gallego Delgado, María
- 5005-2 - ¿CUÁNDO SOLICITAR UN ESTUDIO GENÉTICO EN LA MIOCARDIOPATÍA DILATADA? PREDICTORES DE UN RESULTADO GENÉTICO POSITIVO
- 5005-6 - NUEVA MUTACIÓN EN EL GEN HCN4 EN UNA FAMILIA CON BRADICARDIA SINUSAL COMBINADA CON NO COMPACTACIÓN DEL VENTRÍCULO IZQUIERDO
- 7002-7 - MINOCA: CARACTERIZACIÓN CLÍNICA Y PAPEL DE LA RESONANCIA MAGNÉTICA CARDIACA EN EL DIAGNÓSTICO ETIOLÓGICO DEFINITIVO
- 7004-13 - DESCOMPENSACIÓN DE INSUFICIENCIA CARDIACA POSCARDIOVERSIÓN ELÉCTRICA EN PACIENTES CON FIBRILACIÓN AURICULAR: INCIDENCIA Y PREDICTORES
- García Berrocal, Belén
- Isidoro García, María
-
López Serna, Marta
- 6032-286 - TOMOGRAFÍA COMPUTARIZADA CARDIOVASCULAR GESTIONADA POR CARDIOLOGÍA Y SU PAPEL DENTRO DE UN PROTOCOLO DE UNIDAD DE DOLOR TORÁCICO
- 5005-2 - ¿CUÁNDO SOLICITAR UN ESTUDIO GENÉTICO EN LA MIOCARDIOPATÍA DILATADA? PREDICTORES DE UN RESULTADO GENÉTICO POSITIVO
- 6016-171 - MORTALIDAD A MUY LARGO PLAZO (20 AÑOS) SEGÚN TIPO DE SÍNDROME CORONARIO AGUDO
- 6037-337 - SEGUIMIENTO A LARGO PLAZO DE PACIENTES CON ESTENOSIS AÓRTICA GRAVE TRAS IMPLANTE DE IMPLANTE VALVULAR AÓRTICO TRANSCATÉTER
- 4001-9 - FACTORES RELACIONADOS CON EL DESTETE EXITOSO DE OXIGENADOR CON MEMBRANA EXTRACORPÓREA VENOARTERIAL. EXPERIENCIA EN UN HOSPITAL TERCIARIO
- 6056-489 - ESTRATIFICACIÓN PRONÓSTICA DE LA CLASIFICACIÓN DE KILLIP. ¿SIGUE SIENDO ÚTIL?
- 4003-2 - 25 AÑOS DE SÍNDROME CORONARIO AGUDO Y SHOCK CARDIOGÉNICO ¿CÓMO HEMOS CAMBIADO?
- 4003-7 - EVALUACIÓN DE PARÁMETROS ECOCARDIOGRÁFICOS PREDICTORES EN EL DESTETE DE OXIGENADOR EXTRACORPÓREO DE MEMBRANA VENOARTERIAL
- 7002-7 - MINOCA: CARACTERIZACIÓN CLÍNICA Y PAPEL DE LA RESONANCIA MAGNÉTICA CARDIACA EN EL DIAGNÓSTICO ETIOLÓGICO DEFINITIVO
- Marcos Vadillo, Elena
-
Martín García, Ana
- 6009-76 - EVALUACIÓN ECOCARDIOGRÁFICA BASAL EN ENFERMOS HEMATOLÓGICOS CANDIDATOS A TRASPLANTE ALOGÉNICO DE PRECURSORES HEMATOPOYÉTICOS
- 4031-4 - ANÁLISIS DE LOS BIOMARCADORES CARDIACOS EN EL DIAGNÓSTICO DE SIDEROSIS CARDIACA Y SU VALOR PRONÓSTICO EN EL SÍNDROME MIELODISPLÁSICO DE BAJO GRADO RIESGO
- 7002-7 - MINOCA: CARACTERIZACIÓN CLÍNICA Y PAPEL DE LA RESONANCIA MAGNÉTICA CARDIACA EN EL DIAGNÓSTICO ETIOLÓGICO DEFINITIVO
- 5005-2 - ¿CUÁNDO SOLICITAR UN ESTUDIO GENÉTICO EN LA MIOCARDIOPATÍA DILATADA? PREDICTORES DE UN RESULTADO GENÉTICO POSITIVO
- 7006-7 - ARTERIAS CORONARIAS CON ORIGEN ANÓMALO EN SENO CONTRALATERAL: IMPACTO CLÍNICO Y PRONÓSTICO
- 6032-286 - TOMOGRAFÍA COMPUTARIZADA CARDIOVASCULAR GESTIONADA POR CARDIOLOGÍA Y SU PAPEL DENTRO DE UN PROTOCOLO DE UNIDAD DE DOLOR TORÁCICO
- 4031-7 - ¿ES EL SACUBITRILO/VALSARTÁN ÚTIL EN PACIENTES CON CÁNCER E INSUFICIENCIA CARDIACA? DATOS DEL REGISTRO MULTICÉNTRICO NACIONAL COH-IC
- 5019-8 - REMODELADO VENTRICULAR TRAS EL USO DE SACUBITRILO/VALSARTÁN EN LA MIOCARDIOPATÍA TÓXICA EN EL PACIENTE CON CÁNCER
- 5027-4 - IMPACTO DE LA REDUCCIÓN DE LA SOBRECARGA VOLUMÉTRICA VENTRICULAR DERECHA SOBRE LA INSUFICIENCIA TRICUSPÍDEA FUNCIONAL EN ADULTOS CON CARDIOPATÍAS CONGÉNITAS: SEGUIMIENTO A 6 Y 12 MESES
- 5019-5 - ANÁLISIS DE LA DEFORMACIÓN MIOCÁRDICA COMO FACTOR PREDICTOR PRONÓSTICO DE EVENTOS CARDIOVASCULARES EN EL TRASPLANTE DE PROGENITORES HEMATOPOYÉTICOS
- 4031-6 - SÍNDROME MIELODISPLÁSICO BAJO SOPORTE TRANSFUSIONAL: NUEVA ESCALA PREDICTIVA DE MUERTE Y EVENTOS CARDIOVASCULARES
-
Rodríguez Estévez, Lucía
- 6032-286 - TOMOGRAFÍA COMPUTARIZADA CARDIOVASCULAR GESTIONADA POR CARDIOLOGÍA Y SU PAPEL DENTRO DE UN PROTOCOLO DE UNIDAD DE DOLOR TORÁCICO
- 6051-445 - COMPARACIÓN ENTRE LAS DIFERENTES ETIOLOGÍAS DE LA MIOCARDIOPATÍA DILATADA NO ISQUÉMICA EN UNA COHORTE RETROSPECTIVA DE PACIENTES UTILIZANDO RESONANCIA MAGNÉTICA CARDIACA
- 5005-2 - ¿CUÁNDO SOLICITAR UN ESTUDIO GENÉTICO EN LA MIOCARDIOPATÍA DILATADA? PREDICTORES DE UN RESULTADO GENÉTICO POSITIVO
- 4031-6 - SÍNDROME MIELODISPLÁSICO BAJO SOPORTE TRANSFUSIONAL: NUEVA ESCALA PREDICTIVA DE MUERTE Y EVENTOS CARDIOVASCULARES
- 4001-9 - FACTORES RELACIONADOS CON EL DESTETE EXITOSO DE OXIGENADOR CON MEMBRANA EXTRACORPÓREA VENOARTERIAL. EXPERIENCIA EN UN HOSPITAL TERCIARIO
- 6037-337 - SEGUIMIENTO A LARGO PLAZO DE PACIENTES CON ESTENOSIS AÓRTICA GRAVE TRAS IMPLANTE DE IMPLANTE VALVULAR AÓRTICO TRANSCATÉTER
- 7002-7 - MINOCA: CARACTERIZACIÓN CLÍNICA Y PAPEL DE LA RESONANCIA MAGNÉTICA CARDIACA EN EL DIAGNÓSTICO ETIOLÓGICO DEFINITIVO
- 4003-2 - 25 AÑOS DE SÍNDROME CORONARIO AGUDO Y SHOCK CARDIOGÉNICO ¿CÓMO HEMOS CAMBIADO?
- 4003-7 - EVALUACIÓN DE PARÁMETROS ECOCARDIOGRÁFICOS PREDICTORES EN EL DESTETE DE OXIGENADOR EXTRACORPÓREO DE MEMBRANA VENOARTERIAL
- 6016-171 - MORTALIDAD A MUY LARGO PLAZO (20 AÑOS) SEGÚN TIPO DE SÍNDROME CORONARIO AGUDO
-
Sánchez Fernández, Pedro Luis
- 6032-286 - TOMOGRAFÍA COMPUTARIZADA CARDIOVASCULAR GESTIONADA POR CARDIOLOGÍA Y SU PAPEL DENTRO DE UN PROTOCOLO DE UNIDAD DE DOLOR TORÁCICO
- 5002-8 - ¿ES NECESARIO ANTICOAGULAR LA FIBRILACIÓN AURICULAR DE NOVO EN EL SÍNDROME CORONARIO AGUDO? ESTUDIO OBSERVACIONAL PROSPECTIVO A 20 AÑOS
- 6046-393 - ABLACIÓN DE SUSTRATO EN TORMENTA ARRÍTMICA BAJO SOPORTE CON ASISTENCIA CIRCULATORIA TIPO OXIGENADOR EXTRACORPÓREO DE MEMBRANA
- 6051-445 - COMPARACIÓN ENTRE LAS DIFERENTES ETIOLOGÍAS DE LA MIOCARDIOPATÍA DILATADA NO ISQUÉMICA EN UNA COHORTE RETROSPECTIVA DE PACIENTES UTILIZANDO RESONANCIA MAGNÉTICA CARDIACA
- 5019-5 - ANÁLISIS DE LA DEFORMACIÓN MIOCÁRDICA COMO FACTOR PREDICTOR PRONÓSTICO DE EVENTOS CARDIOVASCULARES EN EL TRASPLANTE DE PROGENITORES HEMATOPOYÉTICOS
- 6009-76 - EVALUACIÓN ECOCARDIOGRÁFICA BASAL EN ENFERMOS HEMATOLÓGICOS CANDIDATOS A TRASPLANTE ALOGÉNICO DE PRECURSORES HEMATOPOYÉTICOS
- 4031-4 - ANÁLISIS DE LOS BIOMARCADORES CARDIACOS EN EL DIAGNÓSTICO DE SIDEROSIS CARDIACA Y SU VALOR PRONÓSTICO EN EL SÍNDROME MIELODISPLÁSICO DE BAJO GRADO RIESGO
- 5005-2 - ¿CUÁNDO SOLICITAR UN ESTUDIO GENÉTICO EN LA MIOCARDIOPATÍA DILATADA? PREDICTORES DE UN RESULTADO GENÉTICO POSITIVO
- 5005-6 - NUEVA MUTACIÓN EN EL GEN HCN4 EN UNA FAMILIA CON BRADICARDIA SINUSAL COMBINADA CON NO COMPACTACIÓN DEL VENTRÍCULO IZQUIERDO
- 4031-6 - SÍNDROME MIELODISPLÁSICO BAJO SOPORTE TRANSFUSIONAL: NUEVA ESCALA PREDICTIVA DE MUERTE Y EVENTOS CARDIOVASCULARES
- 4031-7 - ¿ES EL SACUBITRILO/VALSARTÁN ÚTIL EN PACIENTES CON CÁNCER E INSUFICIENCIA CARDIACA? DATOS DEL REGISTRO MULTICÉNTRICO NACIONAL COH-IC
- 5019-8 - REMODELADO VENTRICULAR TRAS EL USO DE SACUBITRILO/VALSARTÁN EN LA MIOCARDIOPATÍA TÓXICA EN EL PACIENTE CON CÁNCER
- 5006-4 - ESTIMULACIÓN ANTITAQUICARDIA EN TV LENTAS: EFICACIA COMPARADA, PREDICTORES Y CONSECUENCIAS CLÍNICAS DE LA PROGRAMACIÓN DE TRATAMIENTOS SUCESIVOS
- 6016-184 - NOVEDADES EN EL ABORDAJE Y EVOLUCIÓN DEL SÍNDROME CORONARIO AGUDO COMPLICADO KILLIP III Y IV EN LOS ÚLTIMOS 25 AÑOS
- 4017-2 - ¿ES REALMENTE LA FIBRILACIÓN AURICULAR UN FACTOR DE MAL PRONÓSTICO EN EL SÍNDROME CORONARIO AGUDO? ESTUDIO OBSERVACIONAL PROSPECTIVO A 20 AÑOS
- 6003-55 - HEMORRAGIA INTRACRANEAL Y CIERRE PERCUTÁNEO DE LA OREJUELA IZQUIERDA ¿CUÁNDO HACERLO?
- 6031-280 - RELACIÓN DE LAS MEDIDAS PARAMÉTRICAS CON EL CUADRO CLÍNICO DE PRESENTACIÓN EN PACIENTES CON MIOCARDITIS DIAGNOSTICADOS POR RESONANCIA MAGNÉTICA CARDIACA
- 6016-171 - MORTALIDAD A MUY LARGO PLAZO (20 AÑOS) SEGÚN TIPO DE SÍNDROME CORONARIO AGUDO
- 6037-337 - SEGUIMIENTO A LARGO PLAZO DE PACIENTES CON ESTENOSIS AÓRTICA GRAVE TRAS IMPLANTE DE IMPLANTE VALVULAR AÓRTICO TRANSCATÉTER
- 5020-8 - CORRELACIÓN ENTRE LA ESCALA CALCIO DE LA VÁLVULA AÓRTICA Y LA PRESENCIA DE REGURGITACIÓN PARAVALVULAR TRAS EL IMPLANTE DE PRÓTESIS AÓRTICA PERCUTÁNEA
- 5027-4 - IMPACTO DE LA REDUCCIÓN DE LA SOBRECARGA VOLUMÉTRICA VENTRICULAR DERECHA SOBRE LA INSUFICIENCIA TRICUSPÍDEA FUNCIONAL EN ADULTOS CON CARDIOPATÍAS CONGÉNITAS: SEGUIMIENTO A 6 Y 12 MESES
- 7002-7 - MINOCA: CARACTERIZACIÓN CLÍNICA Y PAPEL DE LA RESONANCIA MAGNÉTICA CARDIACA EN EL DIAGNÓSTICO ETIOLÓGICO DEFINITIVO
- 6056-489 - ESTRATIFICACIÓN PRONÓSTICA DE LA CLASIFICACIÓN DE KILLIP. ¿SIGUE SIENDO ÚTIL?
- 6036-307 - HEMORRAGIA INTRACRANEAL Y CIERRE PERCUTÁNEO DE OREJUELA IZQUIERDA, ALGO MÁS QUE UNA ALTERNATIVA
- 4003-2 - 25 AÑOS DE SÍNDROME CORONARIO AGUDO Y SHOCK CARDIOGÉNICO ¿CÓMO HEMOS CAMBIADO?
- 4003-7 - EVALUACIÓN DE PARÁMETROS ECOCARDIOGRÁFICOS PREDICTORES EN EL DESTETE DE OXIGENADOR EXTRACORPÓREO DE MEMBRANA VENOARTERIAL
- 7004-3 - ABLACIÓN CON CATÉTER EN LA TORMENTA ARRÍTMICA ASOCIADA A CICATRIZ: ¿CUÁNTO DEBEMOS ESPERAR?
- 4001-9 - FACTORES RELACIONADOS CON EL DESTETE EXITOSO DE OXIGENADOR CON MEMBRANA EXTRACORPÓREA VENOARTERIAL. EXPERIENCIA EN UN HOSPITAL TERCIARIO
- 7004-13 - DESCOMPENSACIÓN DE INSUFICIENCIA CARDIACA POSCARDIOVERSIÓN ELÉCTRICA EN PACIENTES CON FIBRILACIÓN AURICULAR: INCIDENCIA Y PREDICTORES
- 5018-5 - PREVALENCIA DE ENFERMEDAD CORONARIA SILENTE EN SUJETOS REMITIDOS PARA ABLACIÓN PERCUTÁNEA DE FIBRILACIÓN AURICULAR
- 6038-339 - EFICACIA Y SEGURIDAD DE UNA ESTRATEGIA DE ABORDAJE RADIAL EN EL INTERVENCIONISMO CORONARIO PERCUTÁNEO DE OCLUSIONES TOTALES CRÓNICAS
- 6010-99 - APIXABÁN FRENTE A ACENOCUMAROL EN LA CARDIOVERSIÓN ELÉCTRICA DE PACIENTES CON FIBRILACIÓN AURICULAR: DEL EMANATE A LA VIDA REAL
- 5007-7 - INTELIGENCIA ARTIFICIAL FRENTE A ESTADÍSTICA CLÁSICA EN LA PREDICCIÓN DE CARDIOVERSIÓN ESPONTÁNEA, FARMACOLÓGICA, ELÉCTRICA Y RECURRENCIA DE FIBRILACIÓN AURICULAR
- 7006-7 - ARTERIAS CORONARIAS CON ORIGEN ANÓMALO EN SENO CONTRALATERAL: IMPACTO CLÍNICO Y PRONÓSTICO
-
Villacorta Argüelles, Eduardo
- 5008-6 - ENDOCARDITIS INFECCIOSA: EDUCACIÓN SANITARIA DIRIGIDA A PACIENTES CONSIDERADOS DE ALTO RIESGO
- 5005-2 - ¿CUÁNDO SOLICITAR UN ESTUDIO GENÉTICO EN LA MIOCARDIOPATÍA DILATADA? PREDICTORES DE UN RESULTADO GENÉTICO POSITIVO
- 5001-14 - PREDICTORES DE EMBOLIAS SISTÉMICAS EN NO COMPACTACIÓN DEL VENTRÍCULO IZQUIERDO
- 7002-7 - MINOCA: CARACTERIZACIÓN CLÍNICA Y PAPEL DE LA RESONANCIA MAGNÉTICA CARDIACA EN EL DIAGNÓSTICO ETIOLÓGICO DEFINITIVO
- 5026-2 - PRONÓSTICO DE PACIENTES CON NO COMPACTACIÓN DEL VENTRÍCULO IZQUIERDO Y FRACCIÓN DE EYECCIÓN CONSERVADA
- 5005-6 - NUEVA MUTACIÓN EN EL GEN HCN4 EN UNA FAMILIA CON BRADICARDIA SINUSAL COMBINADA CON NO COMPACTACIÓN DEL VENTRÍCULO IZQUIERDO