Volumen 78. Número 6 (Junio 2025)

Factor de impacto 2024
4,9
ISSN: 0300-8932
Artículos originales
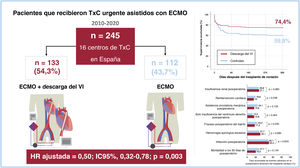
- Impacto de la descarga del ventrículo izquierdo en el resultado tras el trasplante en pacientes asistidos con ECMO-VA
- Daniel Enríquez-Vázquez, Eduardo Barge-Caballero, Francisco González-Vílchez, Luis Almenar-Bonet, María Dolores García-Cosío Carmena, José González-Costello, Manuel Gómez-Bueno, María Ángeles Castel-Lavilla, Beatriz Díaz-Molina, Manuel Martínez-Sellés, Sonia Mirabet-Pérez, Luis de la Fuente-Galán, Daniela Hervás-Sotomayor, Diego Rangel-Sousa, Iris P. Garrido-Bravo, Teresa Blasco-Peiró, Gregorio Rábago Juan-Aracil, Javier Muñiz, María G. Crespo-Leiro
- Rev Esp Cardiol. 2025;78:494-503
- ECMO-VA y descarga ventricular. ¿Una estrategia que puede marcar la diferencia o solo un rayo de esperanza?
- Aitor Uribarri, Eduard Ródenas-Alesina, Ignacio Ferreira-González
- Rev Esp Cardiol. 2025;78:504-6
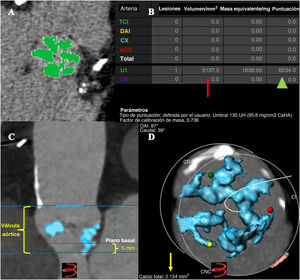
- Volumen de calcificación de la válvula aórtica y su pronóstico en pacientes sometidos a implante percutáneo de válvula aórtica
- Héctor A. Álvarez-Covarrubias, Niklas Altaner, Rafael Adolf, Martin Jurisic, Elisabeth Horban, Costanza Pellegrini, Charlotte Duesmann, Mark Lachmann, Christian Thilo, Finn Syryca, Markus Klos, N. Patrick Mayr, Tobias Rheude, Matthias Renker, Efstratios I. Charitos, Heribert Schunkert, Adnan Kastrati, Erion Xhepa, Kim Won-Keun, Michael Joner
- Rev Esp Cardiol. 2025;78:507-18
- Impacto de la calcificación valvular aórtica en el TAVI. ¿Debemos dar la vuelta a los conceptos previos?
- Alberto Alperi, Raquel del Valle, Pablo Avanzas
- Rev Esp Cardiol. 2025;78:519-20
- Validación experimental de la gravedad de la estenosis coronaria y el desarrollo de miocardio isquémico
- Joo Myung Lee, Seung Hun Lee, Woochan Kwon, Han Byul Kim, David Hong, Hyun Kuk Kim, Sang-Geon Cho, Doosup Shin, Ki Seong Park, Jahae Kim, Jang Bae Moon, Ho-Chun Song, Seungrok Lee, Dong-Heon Ha, Jinah Jang, Youngkeun Ahn, Myung Ho Jeong, Ki Hong Choi, Taek Kyu Park, Jeong Hoon Yang, Young Bin Song, Joo-Yong Hahn, Seung-Hyuk Choi, Hyeon-Cheol Gwon, Young Joon Hong
- Rev Esp Cardiol. 2025;78:521-32
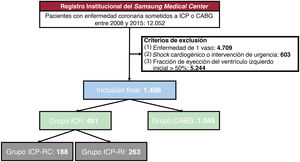
- Resultados tras una intervención coronaria percutánea o cirugía de bypass por miocardiopatía isquémica
- Woochan Kwon, Onyou Kim, Ki Hong Choi, Dong Seop Jeong, Sang Yoon Lee, Joo Myung Lee, Taek Kyu Park, Jeong Hoon Yang, Joo-Yong Hahn, Seung-Hyuk Choi, Su Ryeun Chung, Yang Hyun Cho, Kiick Sung, Wook Sung Kim, Hyeon-Cheol Gwon, Young Tak Lee, Young Bin Song
- Rev Esp Cardiol. 2025;78:533-41
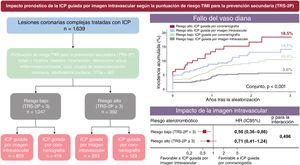
- Impacto pronóstico de la imagen intravascular en la intervención coronaria percutánea según el riesgo aterotrombótico: análisis post hoc de un ensayo clínico aleatorizado
- David Hong, Junho Ha, Ki Hong Choi, Seung Hun Lee, Doosup Shin, Jong-Young Lee, Seung-Jae Lee, Sang Yeub Lee, Sang Min Kim, Kyeong Ho Yun, Jae Young Cho, Chan Joon Kim, Hyo-Suk Ahn, Chang-Wook Nam, Hyuck-Jun Yoon, Yong Hwan Park, Wang Soo Lee, Jeong Hoon Yang, Seung-Hyuk Choi, Hyeon-Cheol Gwon, Young Bin Song, Joo-Yong Hahn, Taek Kyu Park, Joo Myung Lee, Investigators RENOVATE-COMPLEX-PCI
- Rev Esp Cardiol. 2025;78:542-52
- Adherencia a las recomendaciones dietéticas saludables y sostenibles para población española, y mortalidad por todas las causas
- Verónica Vega-Cabello, Almudena Rollán, Isabel Peña-Rey, José R. Banegas, Fernando Rodríguez-Artalejo, Pilar Guallar-Castillón, Esther López-García
- Rev Esp Cardiol. 2025;78:553-62
Miocardiopatías e insuficiencia cardiaca
Comentario editorial
Enfermedad valvular
Comentario editorial
Cardiopatía isquémica y cuidados agudos cardiovasculares

Epidemiología, factores de riesgo y prevención

Articles in press
- Recurrencia rápida de reestenosis coronaria inexplicada en el stent (RECUR): una nueva enfermedad inflamatoria coronaria con evidencia patológica
- Zhangyu Lin, Xuejing Duan, Yuetang Wang, Qian Wang, Lei Jia, Kefei Dou
- 10.1016/j.recesp.2025.04.014
- Disponible online: 18 junio 2025
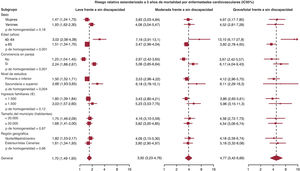
- Asociación de la discapacidad con la mortalidad cardiovascular en la población adulta española no institucionalizada
- Roberto Pastor-Barriuso, Alicia Padrón-Monedero, Fernando J. García López, Javier Almazán-Isla, Jesús de Pedro-Cuesta, Javier Damián
- 10.1016/j.recesp.2025.03.010
- Disponible online: 16 junio 2025
- Abordaje y consumo de recursos sanitarios en la miocardiopatía hipertrófica obstructiva en España: un estudio de la vida real
- Roberto Barriales-Villa, Luis Escobar-López, David Vilanova Larena, Joel Salazar-Mendiguchía, Ainara Echeto, Ignacio Hernández, Elena Rebollo-Gómez, Juan Ramón Gimeno
- 10.1016/j.recesp.2025.04.004
- Pruebas preliminares. Disponible online: 12 junio 2025

Revista Española de Cardiología 75 años en el corazón de la investigación cardiovascular
El coloquio « Revista Española de Cardiología : 75 años en el corazón de la investigación cardiovascular» rememora los logros, dificultades y desafíos de su recorrido único a través de los editores jefe desde los años 90.
Juan Sanchis
Editor jefe de Revista Española de Cardiología (2015-2021-)
Francisco Fernández-Avilés
Editor jefe de Revista Española de Cardiología (1991-1997)
Xavier Bosch
Editor jefe de Revista Española de Cardiologíaz (1997-2003)
Fernando Alfonso
Editor jefe de Revista Española de Cardiología (2004-2009)
Ignacio Ferreira
Editor jefe de Revista Española de Cardiología (2015-2021)