ISSN: 0300-8932
Factor de impacto 2024
4,9
SEC 2021 - El Congreso de la Salud Cardiovascular

Zaragoza,
28 - 30 de Octubre de 2021
Introducción
Dr. Héctor Bueno
Presidente del Comité Científico del Congreso
Comité ejecutivo
Comité de evaluadores
Listado de sesiones
Índice de autores
6040. Cardiopatías familiares y genética cardiovascular II
Fecha
: 30-10-2021 12:00:00
Tipo
: e-póster
Sala
: e-póster 1
6040-13. CORRELACIÓN GENOTIPO-FENOTIPO EN MIOCARDIOPATÍA HIPERTRÓFICA: NUEVA MUTACIÓN P.ARG652LYS EN MYH7
Guido Antoniutti1, Tomás Ripoll Vera1, Jorge Álvarez Rubio1, Paula Morlanes Gracia2, Jaume Pons Llinares3, Elena Fortuny Frau3 y Blanca Rodríguez Picón4
1Hospital Universitario Son Llàtzer, Palma de Mallorca, Illes Balears. 2Hospital Clínico Universitario Lozano Blesa, Zaragoza. 3Hospital Son Espases, Palma de Mallorca, Illes Balears. 4Hospital Mateu Orfila, Menorca, Illes Balears.
1Hospital Universitario Son Llàtzer, Palma de Mallorca, Illes Balears. 2Hospital Clínico Universitario Lozano Blesa, Zaragoza. 3Hospital Son Espases, Palma de Mallorca, Illes Balears. 4Hospital Mateu Orfila, Menorca, Illes Balears.
Introducción y objetivos: La miocardiopatía hipertrófica (MCH) es una enfermedad genética producida por mutaciones en genes sarcoméricos. El gen de la cadena pesada betamiosina (MYH7) es uno de los más prevalentes. Hemos detectado en nuestra región una nueva variante patogénica en este gen, no descrita previamente. Nuestro objetivo fue establecer la correlación genotipo-fenotipo de la variante p.Arg652Lys en una serie de familias.
Métodos: Estudio descriptivo de pacientes con diagnóstico de MCH y hallazgo de dicha variante mediante técnica Sanger o NGS. Se analizó la cosegregación, las características clínicas, de imagen (electrocardiograma, ecocardiograma, resonancia cardiaca, ergometría y holter) y los eventos cardiovasculares.
Resultados: Se identificó la variante p.Arg652Lys en MHY7 en 8 familias. De un total de 59 pacientes estudiados, 39 (66%) presentaron la mutación, con una mediana de edad al inicio del estudio de 39 años y una mediana de seguimiento de 63 meses. Se observó desarrollo de MCH en 25 (64%) portadores de la mutación. Dentro de los pacientes con fenotipo de MCH se observó una mediana de espesor parietal del ventrículo izquierdo de 16,5 mm (RIQ [25-75] 13,8-22 mm), función sistólica conservada en todos los pacientes, fibrilación auricular en 3 (12%) pacientes, gradiente obstructivo > 30 mmHg en 7 (28%), realce tardío en resonancia cardiaca en 5 (20%), sin registros de extensa fibrosis. No se observó desarrollo de cardiopatía en pacientes no portadores de la mutación. Se analizó en un compuesto de eventos clínicos adversos (ECA) que incluyó muerte súbita (MS), muerte súbita abortada (MSA), descarga apropiada de desfibrilador, evento embólico (ictus o evento isquémico periférico) e ingreso por insuficiencia cardiaca. Dicho compuesto se presentó en 5 (20%) pacientes, 1 (4%) presentó MS, 1 (4%) MSA y 3 (12%) eventos embólicos. Siendo únicamente el evento de MSA en un paciente menor de 50 años de edad (48 años).
|
Incidencia de eventos clínicos en pacientes con hallazgo de la mutación p.Arg652Lys en MYH7 |
|||
|
Eventos clínicos |
|||
|
ECA (N -%) |
5 |
20,0% |
|
|
Muerte súbita (N -%) |
1 |
4,0% |
|
|
MS en menores de 50 años de edad (N -%) |
0 |
0,0% |
|
|
MS recuperada (N -%) |
1 |
4,0% |
|
|
Descarga apropiada de DAI (N -%) |
0 |
0,0% |
|
|
IC (N -%) |
2 |
8,0% |
|
|
Ingreso por IC (N -%) |
0 |
0,0% |
|
|
FA (N -%) |
3 |
12,0% |
|
|
Evento embólico (ictus/isquemia periférica) (N -%) |
3 |
12,0% |
|
|
TRS (N -%) |
1 |
4,0% |
|
|
Ablación con alcohol (N -%) |
0 |
0,0% |
|
|
Miectomía (N -%) |
1 |
4,0% |
|
|
DAI: desfribrilador implantable; ECA: evento clínico adverso; FA: fibrilación auricular; IC: insuficiencia cardiaca; MS: muerte súbita; RIQ: rango intercuartil; TRS: terapia de reducción septal. |
|||
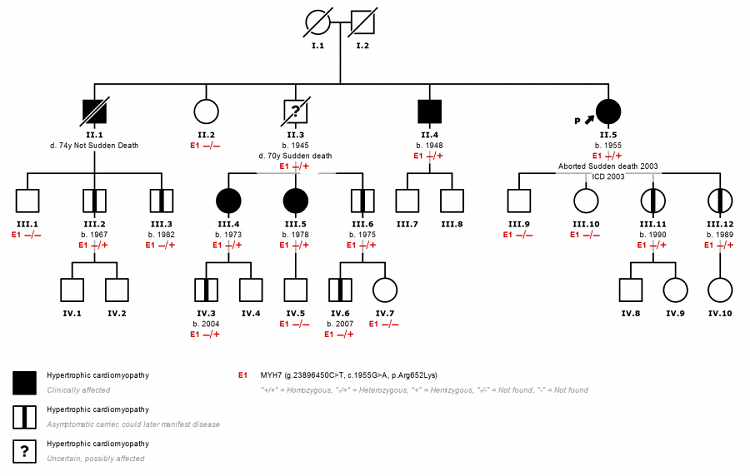
Árbol genealógico de una de las familias con la mutación p.Arg652Lys en MHY7.
Conclusiones: Hemos demostrado la patogenicidad de la variante p.Arg652Lys en MHY7 tras estudio de cosegregación en nuestras familias. La misma está asociada con MCH de penetrancia incompleta y moderado riesgo clínico aunque con presentación de ECA generalmente a edad avanzada. El hecho de presentarse en 8 familias de la región y no hallarse descripción previa de la misma en otra localización sugiere un efecto fundador.
Comunicaciones disponibles de "Cardiopatías familiares y genética cardiovascular II"
- 6040-1. MODERADORA
- Carmen Muñoz Esparza, Murcia
- 6040-2. DINÁMICA VALVULAR PULMONAR TRAS CORRECCIÓN DE TETRALOGÍA DE FALLOT
- Francisco Javier Soria Romero, María Inmaculada Navarrete Espinosa, Miriam Jiménez González, Lourdes Conejo Muñoz y Cristóbal Urbano Carrillo
Hospital Regional Universitario Carlos Haya, Málaga.
- 6040-3. SÍNDROMES AÓRTICOS AGUDOS EN JÓVENES ¡RED FLAG! SOSPECHA AORTOPATÍA GENÉTICA
- Nerea Mora Ayestarán, Mayte Basurte Elorz, Ignacio Roy Añon, Gemma Lacuey Lecumberri, Aitor Ansotegui Hernández, Aitziber Munarriz Arizcuren, Javier Martínez Basterra, Marina Oliver Ledesma, Betel Olaizola Balboa y Virginia Álvarez Asiain
Complejo Hospitalario de Navarra, Pamplona/Iruña, Navarra.
- 6040-4. LA DISPERSIÓN MECÁNICA MEDIDA POR STRAIN ES CAPAZ DE IDENTIFICAR A LOS PORTADORES DE LA VARIANTE GENÉTICA EN FAMILIARES DE PACIENTES CON MIOCARDIOPATÍA HIPERTRÓFICA
- Carlos Gutiérrez Landaluce, Iria Andrea González García, Carmen Cristóbal Varela, José María Serrano Antolín, Rosa Mª Jiménez Hernández, Alejandro Curcio Ruigómez, Pedro Luis Talavera Calle, Silvia del Castillo Arrojo, Catherine Graupner Abad, Adriana de la Rosa Riestra, Javier Alonso Belló y Elena Magallanes-Ribeiro Catalán
Hospital Universitario de Fuenlabrada, Madrid.
- 6040-5. RELACIÓN DEL NIVEL DE ACTIVIDAD FÍSICA Y EL CURSO EVOLUTIVO DE LA ENFERMEDAD EN PACIENTES CON SÍNDROME DE BRUGADA
- David Fernández Vázquez1, María Jesús Fernández Gil1, Lidia María Carrillo Mora1, Francisco Melgarejo Meseguer2, Agustín Ramos López1, Juan José Santos Mateo3, Carmen Muñoz Esparza1, Ana Isabel Rodríguez Serrano4, Marina Navarro Peñalver5, Noelia Fernández Villa1, Eva Cabrera Romero1, Francisco Javier Gimeno Blanes2 y Juan Ramón Gimeno Blanes1
1Hospital Clínico Universitario Virgen de la Arrixaca, Murcia. 2Universidad Miguel Hernández, Elche, Alicante. 3Hospital Rafael Méndez, Lorca, Murcia. 4Hospital Universitario J.M. Morales Meseguer, Murcia. 5Hospital Comarcal del Noroeste, Caravaca de la Cruz, Murcia.
- 6040-6. UTILIDAD DEL SCREENING FAMILIAR EN EL ANEURISMA AÓRTICO FAMILIAR NO SINDRÓMICO
- Julia Rodríguez Ortuño, María Luisa Peña Peña y Alejandro Adsuar Gómez
Hospital Universitario Virgen del Rocío, Sevilla.
- 6040-7. VALOR PRONÓSTICO DEL NT-PROBNP Y ST2 EN PACIENTES CON DIAGNÓSTICO DE MIOCARDIOPATÍA HIPERTRÓFICA
- Helena Resta Saurí, Germán Cediel Calderón, María del Mar Domingo Teixidor, Pau Codina Verdaguer, Giosafat Spitaleri, Evelyn Santiago Vacas, Patricia Velayos Martos, Ana Mª Pulido Altamirano, Eva Crespo García, Carmen Rivas Jiménez, Beatriz González Fernández, Joan F. Andrés Cordón, Andrea Borrellas Martín, Josep Lupón Rosés y Antonio Bayés Genís
Hospital Universitari Germans Trias i Pujol, Badalona, Barcelona.
- 6040-8. LA GENÉTICA COMO AYUDA A LA ENTELEQUIA DE LA NO COMPACTACIÓN MIOCÁRDICA
- María Valverde Gómez, Luis de la Higuera Romero, Soledad García-Hernández, Noel Brogger, Ivonne J. Cárdenas, Germán Fernández Ferro, Xusto Fernández, Arsonval Lamounier, Diego García Giustiniani, Juan Pablo Ochoa, Martín Ortiz Genga y William J. Mckenna
Health in Code, A Coruña.
- 6040-9. NUEVA FUENTE DE CULTIVOS PARA EL ESTUDIO DE CARDIOPATÍAS A PARTIR DE MUESTRAS FORENSES
- Lara Milián1, Aitana Braza-Boils2, María Oliver1, Mar Nieto3, Pilar Molina4, Juan Giner4, Yolanda Abellán4, Jennifer Sancho4, Luis Martínez-Dolz5, Manuel Mata1 y Esther Zorio6
1Unidad de Histología, Facultad de Medicina y Odontología, Universidad de Valencia, 2Grupo Acreditado CaFaMuSME, Fundación de Investigación Hospital Universitario y Politécnico La Fe, CIBERCV, ISCIII, Valencia. 3Unidad de Histología, Facultad de Medicina y Odontología, Universidad de Valencia, Hospital Universitario La Fe, Valencia. 4Instituto de Medicina Legal y Forense, Grupo Acreditado CaFaMuSME, Fundación para la Investigación del Hospital Universitario y Politécnico La Fe, Valencia. 5Servicio de Cardiología, Hospital Universitario La Fe, CIBERCV, ISCIII, Valencia. 6Servicio de Cardiología, Hospital Universitario La Fe, Grupo CaFaMuSME, Fundación Investigación Hospital Universitario y Politécnico La Fe, CIBERCV, ISCIII, Valencia.
- 6040-10. BIOPSIA ENDOMIOCÁRDICA EN PACIENTES CON SOSPECHA DE MIOCARDIOPATÍA INFILTRATIVA
- Cristina Aguilera Agudo, Juan Francisco Oteo Domínguez, Eusebio García-Izquierdo Jaén, Carlos Arellano Serrano, Fernando Domínguez Rodríguez, Arturo García Touchard, José Antonio Fernández Díaz, María del Trigo Espinosa, Francisco Javier Goicolea Ruigómez, Pablo García Pavía, Clara Salas Antón y Javier Segovia Cubero
Hospital Universitario Puerta de Hierro, Majadahonda, Madrid.
- 6040-11. ¿LA PRESENCIA DE PATRÓN DE NO COMPACTACIÓN IMPLICA PEOR PRONÓSTICO EN MIOCARDIOPATÍA DILATADA?
- Nerea Mora Ayestarán, Ignacio Roy Añon, Gonzalo Luis Alonso Salinas, Mayte Basurte Elorz, Gemma Lacuey Lecumberri, Mercedes Ciriza Esandi, Arturo Lanaspa Gallego, Jara Amaiur García Ugaldebere, Julene Ugarriza Ortueta y Virginia Álvarez Asiain
Complejo Hospitalario de Navarra, Pamplona/Iruña, Navarra.
- 6040-12. DIAGNÓSTICO DE ENFERMEDAD DE FABRY EN UN HOSPITAL DE TERCER NIVEL: UN RETO PARA EL CARDIÓLOGO
- Miguel Soroa Ortuño, Elena Astudillo Cortés, Noemi Barja González, Alejandro Junco Vicente, Andrea Aparicio Gavilanes, David Ledesma Olóriz, Javier Martínez Díaz, Pedro Vidau Argüelles, Jesús Mª de la Hera Galarza y Ana Fidalgo Argüelles
Hospital Universitario Central de Asturias, Oviedo, Asturias.
- 6040-13. CORRELACIÓN GENOTIPO-FENOTIPO EN MIOCARDIOPATÍA HIPERTRÓFICA: NUEVA MUTACIÓN P.ARG652LYS EN MYH7
- Guido Antoniutti1, Tomás Ripoll Vera1, Jorge Álvarez Rubio1, Paula Morlanes Gracia2, Jaume Pons Llinares3, Elena Fortuny Frau3 y Blanca Rodríguez Picón4
1Hospital Universitario Son Llàtzer, Palma de Mallorca, Illes Balears. 2Hospital Clínico Universitario Lozano Blesa, Zaragoza. 3Hospital Son Espases, Palma de Mallorca, Illes Balears. 4Hospital Mateu Orfila, Menorca, Illes Balears.
- 6040-14. EVOLUCIÓN A LARGO PLAZO DE PACIENTES CON MIOCARDIOPATÍA HIPERTRÓFICA PORTADORES DE DESFIBRILADOR AUTOMÁTICO IMPLANTABLE EN PREVENCIÓN PRIMARIA Y SECUNDARIA
- Andrea Aparicio Gavilanes1, Nerea González Hompanera2, Diego Pérez Díez1, José Julián Rodríguez Reguero1, María Martín Fernández1, David Ledesma Olóriz1, Javier Martínez Díaz1, Alejandro Junco Vicente1, Miguel Soroa Ortuño1, Noemí Barja González1, Antonio Adeba García1, Rut Álvarez Velasco1, María Vigil-Escalera Díaz1 y Rebeca Lorca Gutiérrez1
1Hospital Universitario Central de Asturias, Oviedo, Asturias. 2Universidad de Oviedo, Asturias.
- 6040-15. PRONÓSTICO Y EVOLUCIÓN DE LA NO COMPACTACIÓN DEL VENTRÍCULO IZQUIERDO. ¿MIOCARDIOPATÍA O VARIANTE DE LA NORMALIDAD?
- Nerea Mora Ayestarán, Gonzalo Luis Alonso Salinas, Ignacio Roy Añon, Mayte Basurte Elorz, Gemma Lacuey Lecumberri, Marina Oliver Ledesma, Julene Ugarriza Ortueta, Jara Amaiur García Ugaldebere, Arturo Lanaspa Gallego y Virginia Álvarez Asiain
Complejo Hospitalario de Navarra, Pamplona/Iruña, Navarra.
- 6040-16. PREDICTORES CLÍNICOS, DE LABORATORIO, ELECTOCARDIOGRÁFICOS Y DE IMAGEN MULTIMODAL EN AMILOIDOSIS CARDIACA
- Rubén Fernández Galera1, Yassin Behnalech1, Ilaria Dentamaro1, Xabier Cia Mendioroz2, Laura Gutiérrez García-Moreno1, Laura Galian Gay1, Guillem Casas Masnou1, Filipa Valente1, Gisela Teixido Tura1, María Luz Servato1, María Isabel González del Hoyo1, M. Teresa González Alujas1 y José Rodríguez-Palomares1
1Hospital Universitario Vall d'Hebron, Barcelona. 2Hospital Puerta de Hierro, Madrid.
- 6040-17. PERFIL CLÍNICO-EPIDEMIOLÓGICO DE LAS ANOMALÍAS CORONARIAS COMO CAUSA DE MUERTE SÚBITA
- José Antonio Sorolla Romero1, Javier Navarrete Navarro1, Pilar Molina Aguilar2, María Paz Suárez Mier3, Joaquín Lucena Romero4, Susana Moyano Corvillo5, Joaquín Rueda Soriano1, Esther Zorio Grima1 y Luis Martínez Dolz1
1Hospital Universitario La Fe, Valencia. 2Instituto de Medicina Legal y Forense, Valencia. 3Instituto Nacional de Toxicología y Ciencias Forenses, Madrid. 4Instituto de Medicina Legal y Ciencias Forenses, Sevilla. 5Instituto Nacional de Toxicología y Ciencias Forenses, Barcelona.
- 6040-18. IMPACTO A LARGO PLAZO DEL BLOQUEO DEL SISTEMA RENINA-ANGIOSTERONA EN EL SÍNDROME DE TAKO-TSUBO
- María Cespón Fernández1, Sergio Raposeiras Roubín1, Emad Abu Assi1, Iván Núñez Gil2, Karim Jamhour-Chelh1, David Aritza Conty3, Óscar Ángel Vedia Cruz2, Manuel Almendro Delia4, Alessandro Sionis Green5, Agustín Carlos Martín García6, Miguel José Corbí Pascual7, Manuel Martínez Sellés8, Aitor Uribarri González9 y Marta Guillén Marzo10
1Hospital Universitario Álvaro Cunqueiro, Vigo; Pontevedra. 2Hospital Clínico San Carlos, Madrid. 3Complejo Hospitalario de Navarra, Pamplona/Iruña, Navarra. 4Hospital Universitario Virgen Macarena, Sevilla. 5Hospital de la Santa Creu i Sant Pau, Barcelona. 6Hospital Clínico Universitario de Salamanca. 7Complejo Hospitalario Universitario de Albacete. 8Hospital General Universitario Gregorio Marañón, Madrid. 9Hospital Clínico Universitario de Valladolid. 10Hospital Universitario Joan XXIII, Tarragona.
Más comunicaciones de los autores
- Álvarez Rubio, Jorge
- Antoniutti, Guido
- Fortuny Frau, Elena
-
Morlanes Gracia, Paula
- 6015-12 - INTERVENCIÓN ANTITABACO PRECOZ DURANTE LA HOSPITALIZACIÓN POR SÍNDROME CORONARIO AGUDO EN PACIENTES INCLUIDOS EN UN PROGRAMA DE REHABILITACIÓN CARDIACA
- 6009-4 - IMPLICACIONES PRONÓSTICAS DE LA PRESENTACIÓN ELECTROCARDIOGRÁFICA DE LOS PACIENTES CON INFARTO AGUDO DE MIOCARDIO Y OCLUSIÓN COMPLETA DE LA ARTERIA RESPONSABLE
- 6031-9 - NUEVA MUTACIÓN EN NKX2.5 EN UNA FAMILIA CON DEFECTOS SEPTALES CONGÉNITOS, MIOCARDIOPATÍA NO COMPACTADA, TRASTORNOS DE LA CONDUCCIÓN Y MUERTE SÚBITA
- 6002-4 - COMPLICACIONES TROMBOEMBÓLICAS Y HEMORRÁGICAS EN EL SEGUIMIENTO A LARGO PLAZO TRAS REEMPLAZO VALVULAR AÓRTICO POR ESTENOSIS AÓRTICA GRAVE
- 6040-13 - CORRELACIÓN GENOTIPO-FENOTIPO EN MIOCARDIOPATÍA HIPERTRÓFICA: NUEVA MUTACIÓN P.ARG652LYS EN MYH7
- Pons Llinares, Jaume
-
Ripoll Vera, Tomás
- 6031-9 - NUEVA MUTACIÓN EN NKX2.5 EN UNA FAMILIA CON DEFECTOS SEPTALES CONGÉNITOS, MIOCARDIOPATÍA NO COMPACTADA, TRASTORNOS DE LA CONDUCCIÓN Y MUERTE SÚBITA
- 4016-7 - PRONÓSTICO DE PACIENTES CON NO COMPACTACIÓN DEL VENTRÍCULO IZQUIERDO Y FUNCIÓN SISTÓLICA CONSERVADA
- 6040-13 - CORRELACIÓN GENOTIPO-FENOTIPO EN MIOCARDIOPATÍA HIPERTRÓFICA: NUEVA MUTACIÓN P.ARG652LYS EN MYH7
- Rodríguez Picón, Blanca