ISSN: 0300-8932
Factor de impacto 2023
7,2
SEC 2024 - El Congreso de la Salud Cardiovascular

Bilbao,
24 - 26 de Octubre de 2024
Introducción
Dr. José María de la Torre Hernández
Presidente del Comité Científico del Congreso. Vicepresidente de la SEC
Comités ejecutivo, organizador y científico
Comité de evaluadores
Listado completo de comunicaciones
Índice de autores
4008. Comunicaciones en cardiogenética, miocardiopatías y aortopatías
Fecha
: 24-10-2024 15:45:00
Tipo
: Comunicaciones orales
Moderadores
: Almudena Amor Salamanca. Área de Genética Cardiovascular, Health In Code, A Coruña
4008-6. ¿Es útil el screening familiar en miocardiopatía hipertrófica?
Cristina Gómez González1, Alexis Rojas1, Silvia Vilches Soria1, Ana Paula Carrillo2, Teresa Bañuelos2, Anaëlle Garoux2, Irene Méndez Fernández1, Reyes Álvarez García-Rovés3, Miriam Centeno Jiménez3, Constancio Medrano López3, Ana Isabel Fernández Ávila4, Renée Olsen Rodríguez5, Nélida Vázquez Aguilera1, Javier Bermejo Thomas1 y M.M. Ángeles Espinosa Castro1
1Cardiología. Hospital General Universitario Gregorio Marañón, Madrid, España, 2Facultad de Medicina. Universidad Complutense, Madrid, España, 3Cardiología Pediátrica. Hospital General Universitario Gregorio Marañón, Madrid, España, 4CIBERCV. Hospital General Universitario Gregorio Marañón, Centro de Investigación Biomédica en Red, Enfermedades Cardiovasculares (CIBERCV), Madrid, España y 5Cardiología. Hospital Universitario de Getafe, Getafe (Madrid), España.
1Cardiología. Hospital General Universitario Gregorio Marañón, Madrid, España, 2Facultad de Medicina. Universidad Complutense, Madrid, España, 3Cardiología Pediátrica. Hospital General Universitario Gregorio Marañón, Madrid, España, 4CIBERCV. Hospital General Universitario Gregorio Marañón, Centro de Investigación Biomédica en Red, Enfermedades Cardiovasculares (CIBERCV), Madrid, España y 5Cardiología. Hospital Universitario de Getafe, Getafe (Madrid), España.
Introducción y objetivos: El screening de familiares de probandos con miocardiopatía hipertrófica (MH) es práctica clínica habitual pero la evidencia acerca del rendimiento diagnóstico es escasa. Dos estudios no relacionados sugieren que hasta un 50% de los portadores podrían desarrollar fenotipo, mientras que muy pocos familiares desarrollan MH cuando en el probando no se identifica una variante causal. No hay comparaciones directas entre ambos grupos y no hay evidencia de la mejor estrategia de screening en familiares con genética desconocida. Nos planteamos determinar la aparición de fenotipo de MH, comparando individuos portadores de variantes causales con aquellos con genética desconocida en el probando y describir la tasa de eventos en el seguimiento.
Métodos: Se incluyeron retrospectivamente 566 familiares de probandos con MH evaluados en una Unidad de Cardiopatías Familiares entre enero de 2017 y abril de 2023. Se comparó la aparición de fenotipo en familiares portadores de una variante causal y familiares con genética no identificada en el probando. Se recogieron los eventos en el seguimiento.
Resultados: La tabla muestra las características basales de los 566 familiares analizados. En 312 casos el screening fue clínico y genético y en 254 solo clínico. El 50% (156/312) de los familiares testados eran portadores. La media de seguimiento fue de 4,05 años. Se diagnosticó de MH a un 26% (40/156) de familiares portadores y a un 6% (15/254) de familiares sin genética conocida. El 25% (14/55) de los casos fueron diagnosticados en la primera consulta. La relación entre desarrollo de fenotipo y ser portador de variante patogénica frente a una genética desconocida resultó significativa [p < 0,0001; OR = 5,5 (IC95% 2,8-11,1)] en el análisis univariado y en el análisis de supervivencia [HR 6,86 (IC95% 3,34-14,09), p < 0,00001] con una separación temprana de las curvas (figura). En el seguimiento hubo escasos eventos en ambos grupos (tabla).
|
Características basales y eventos en seguimiento |
||||
|
Características basales todos los pacientes (N = 566) |
Sujetos del análisis estadístico (N = 410) |
|||
|
Estudio genético positivo (N = 156) |
No estudio genético (N = 254) |
|||
|
Sexo femenino (n, %) |
309 (54,6%) |
161 (48,2%) |
166 (59,9%) |
|
|
Edad en la primera visita (mediana, DE, rango) |
37 años (DE 19,0, 1 mes- 89 años) |
30 años (DE 20,6) |
44 años (DE 14) |
|
|
DM (n, %) |
20 (3,5%) |
8 (5,1%) |
8 (3,2%) |
|
|
HTA (n, %) |
67 (11,8%) |
16 (10,3%) |
39 (15,3%) |
|
|
DL (n, %) |
68 (12,0%) |
15 (9,6%) |
42 (16,5%) |
|
|
Tabaquismo (n, %) |
48 (8,5%) |
8 (5,3%) |
25 (9,8%) |
|
|
Historia cardiológica |
CIC (n, %) |
6 (1,1%) |
1 (0,6%) |
1 (0,4%) |
|
MS previa (n, %) |
1 (0,2%) (disección coronaria) |
0 (0,0%) |
1 (0,4%) |
|
|
FA (n, %) |
6 (1,1%) |
3 (1,9%) |
1 (0,4%) |
|
|
NYHA primera visita (n, %) |
I: 550 (97,3%) |
I: 150 (96,2%) |
I: 247 (96,2%) |
|
|
II: 14 (2,5%) |
II: 5 (3,2%) |
II: 7 (2,8%) |
||
|
III: 1 (0,2%) |
III: 1 (0,6%) |
III: 0 (0%) |
||
|
Síntomas (disnea, dolor torácico, síncope) (n, %) |
24 (4,2%) |
8 (5,1%) |
10 (4,0%) |
|
|
MS familiar < 40 años (n, %) |
63 (11,1%) |
20 (12,9%) |
15 (5,9%) |
|
|
Eventos en el seguimiento en diagnosticados con miocardiopatía hipertrófica (N = 55) |
Portadores variante LP/P (N = 40) |
Genética desconocida (N = 15) |
||
|
ACV (n, %) |
2 (3,6%) |
2 (5%) |
0 (0,0%) |
|
|
FA (n, %) |
1 (1,8%) |
1 (2,5%) |
0 (0,0%) |
|
|
TVMS (n, %) |
1 (1,8%) |
1 (2,5%) |
0 (0,0%) |
|
|
IC (n, %) |
0 (0,0%) |
0 (0,0%) |
0 (0,0%) |
|
|
DAI |
Implante DAI (n, %) |
2 (3,6%) |
1 (2,5%) |
1 (6,7%) |
|
Descargas DAI (n, %) |
0 (0,0%) |
0 (0,0%) |
0 (0,0%) |
|
|
Muerte (n, %) |
0 (0,0%) |
0 (0,0%) |
0 (0,0%) |
|
|
DE: desviación estándar; DM: diabetes mellitus; DL: dislipemia; HTA: hipertensión arterial; CIC: cardiopatía isquémica crónica; MS: muerte súbita; NYHA: Clase funcional de la New York Heart Association; FA: fibrilación auricular; ACV: accidente cerebrovascular; TVMS: taquicardia ventricular monomorfa sostenida; IC: insuficiencia cardiaca; DAI: desfibrilador automático implantable. |
||||
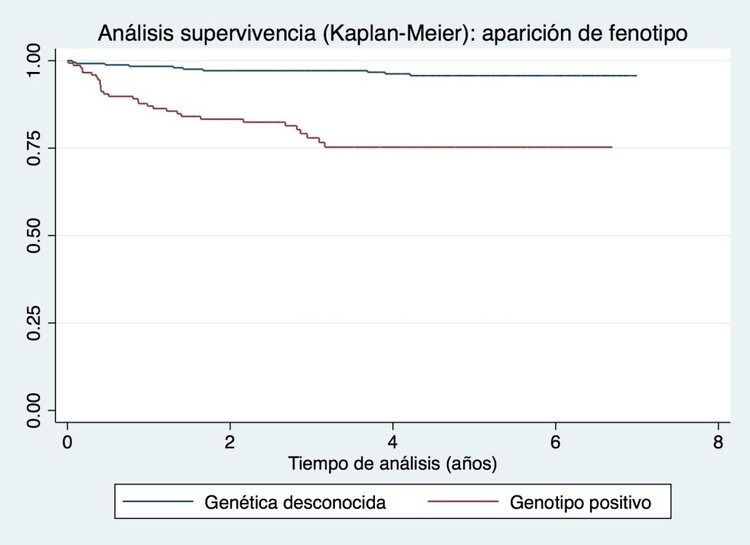
Análisis de supervivencia portadores de variante causal de MH (patogénica o probablemente patogénica) vs genética desconocida.
Conclusiones: En nuestra cohorte el 9,6% de los familiares estudiados presentaron fenotipo aunque los portadores tenían un riesgo muy superior de MH frente a familiares sin genética conocida. La baja probabilidad de fenotipo en estos casos unido a los escasos eventos apoyaría ampliar los intervalos de screening clínico o incluso valorar detenerlo en casos seleccionados cuando no hay genética identificada tras un primer cribado normal.
Comunicaciones disponibles de "4008. Comunicaciones en cardiogenética, miocardiopatías y aortopatías"
- 4008-1. Modera
- Almudena Amor Salamanca. Área de Genética Cardiovascular, Health In Code (A Coruña)
- 4008-2. Caracterización de polimorfismos genéticos del CYP450 asociados a síndrome de QT largo inducido por fármacos en una gran cohorte prospectiva
- Bieito Campos García1, Juliana Salazar Blanco2, Benjamín Rodríguez Santiago3, Héctor Hernández Ontiveros4, Aina Ávila Parcet5, Víctor García Hernando1, Mar Carceller Sindreu5, Ana Juanes Borrego6, María Antonia Martí Femenías7, Enrique Rodríguez Font1, Concepción Alonso Martín1, Zoraida L. Moreno Weidmann1, Francisco Méndez Zurita1, Xavier Viñolas Prat1 y José M.M. Guerra Ramos1
1Servicio de Cardiología. Hospital de la Santa Creu i Sant Pau, Barcelona, España, 2Unidad de Genética. Institut d’Investigació Biomèdica Sant Pau, Barcelona, España, 3Servicio de Genética. Hospital de la Santa Creu i Sant Pau, Barcelona, España, 4Servicio de Medicina Interna. Hospital de la Santa Creu i Sant Pau, Barcelona, España, 5Servicio de Psiquiatría. Hospital de la Santa Creu i Sant Pau, Barcelona, España, 6Servicio de Farmacia. Hospital de la Santa Creu i Sant Pau, Barcelona, España y 7Hospital de la Santa Creu i Sant Pau, Barcelona, España.
- 4008-3. Impacto de variantes sinónimas en las miocardiopatías hereditarias
- Ana Isabel Fernández Ávila1, Silvia Vilches Soria2, Irene Méndez Fernández2, Cristina Gómez González2, Renée Olsen Rodríguez3, Nélida Vázquez Aguilera2, Reyes Álvarez-García Revés4, Miriam Centeno Jiménez4, Constancio Medrano López4, Javier Bermejo Thomas2 y M.M. Ángeles Espinosa Castro2
1Cardiología. Centro de Investigación Biomédica en Red de Enfermedades Cardiovasculares (CIBER-CV), Madrid, España, 2Cardiología. Hospital General Universitario Gregorio Marañón, Madrid, España, 3Cardiología. Hospital Universitario de Getafe, Getafe (Madrid), España y 4Cardiología Pediátrica. Hospital General Universitario Gregorio Marañón, Madrid, España.
- 4008-4. Predictores de eventos adversos en patología aórtica. No solo el tamaño importa
- Ismael Arco Adamuz1, Miguel Morales García1, Inés Uribe Morales1, Diego Segura Rodríguez2, Eduardo Moreno Escobar2 y Rocío García Orta1
1Hospital Universitario Virgen de las Nieves, Granada, España y 2Hospital Clínico San Cecilio, Granada, España.
- 4008-5. Diferencias de género en el pronóstico aórtico en pacientes con síndrome de Marfan. Estudio longitudinal retrospectivo del registro REPAG
- Gisela Teixido Tura1, Diana Domingo Valero2, Rocío García Orta3, Francisco Calvo Iglesias4, Francisco Valera Martínez2, José María Larrañaga Moreira5, Daniel Martínez López6, Clara Badia Molins1, Rosario Sánchez Martínez7, Anna Sabaté Rotés8, Julián Palomino-Doza9, Javier Limeres Freire1, Eduardo Moreno Escobar3, Alberto Forteza Gil6 y Fernando Cabrera Bueno10
1Hospital Universitari Vall d'Hebron, Centro de Investigación Biomédica en Red, Enfermedades Cardiovasculares (CIBERCV), Barcelona, España, 2Hospital Universitario La Fe, Valencia, España, 3Hospital Universitario Virgen de las Nieves, Granada, España, 4Hospital Álvaro Cunqueiro, Vigo (Pontevedra), España, 5Complexo Hospitalario Universitario A Coruña, A Coruña, España, 6Hospital Universitario Puerta de Hierro, Madrid, España, 7Hospital General Universitario de Alicante, Alicante, España, 8Hospital Universitari Vall d'Hebron, Barcelona, España, 9Hospital Universitario 12 de Octubre, Madrid, España y 10Hospital Clínico Universitario Virgen de la Victoria, Málaga, España.
- 4008-6. ¿Es útil el screening familiar en miocardiopatía hipertrófica?
- Cristina Gómez González1, Alexis Rojas1, Silvia Vilches Soria1, Ana Paula Carrillo2, Teresa Bañuelos2, Anaëlle Garoux2, Irene Méndez Fernández1, Reyes Álvarez García-Rovés3, Miriam Centeno Jiménez3, Constancio Medrano López3, Ana Isabel Fernández Ávila4, Renée Olsen Rodríguez5, Nélida Vázquez Aguilera1, Javier Bermejo Thomas1 y M.M. Ángeles Espinosa Castro1
1Cardiología. Hospital General Universitario Gregorio Marañón, Madrid, España, 2Facultad de Medicina. Universidad Complutense, Madrid, España, 3Cardiología Pediátrica. Hospital General Universitario Gregorio Marañón, Madrid, España, 4CIBERCV. Hospital General Universitario Gregorio Marañón, Centro de Investigación Biomédica en Red, Enfermedades Cardiovasculares (CIBERCV), Madrid, España y 5Cardiología. Hospital Universitario de Getafe, Getafe (Madrid), España.
- 4008-7. Cohorte nacional de sospecha de miocarditis Pre-MYO: manejo asistencial en los 100 primeros casos
- Fernando Domínguez Rodríguez1, Alejandro Riquelme Pérez2, Ángela Carrillo Molina2, José María Larrañaga Moreira3, Jara Gayán Ordás4, Marcelo Sanmartín Fernández5, Belén Álvarez Álvarez6, Javier Torres Llergo7, Héctor Bueno Zamora8, Francisco Ridocci Soriano9, Elena Sufrate Sorzano10, Manuel Barreiro Pérez11, José María Viéitez Flórez12, Beatriz Fernández González13 y Domingo Pascual-Figal14
1Cardiología. Hospital Universitario Puerta de Hierro, Majadahonda (Madrid), España, 2Cardiología. Instituto Murciano de Investigación Biosanitaria Virgen de la Arrixaca, Murcia, España, 3Cardiología. Complexo Hospitalario Universitario A Coruña, A Coruña, España, 4Cardiología. Hospital Universitari Arnau de Vilanova, Lleida, España, 5Cardiología. Hospital Universitario Ramón y Cajal, Madrid, España, 6Cardiología. Complexo Hospitalario Universitario de Santiago de Compostela, Santiago de Compostela (A Coruña), España, 7Cardiología. Hospital Universitario de Jaén, Jaén, España, 8Cardiología. Hospital Universitario 12 de Octubre, Madrid, España, 9Cardiología. Hospital General Universitario, Valencia, España, 10Cardiología. Complejo Hospitalario San Millán-San Pedro, Logroño (La Rioja), España, 11Cardiología. Hospital Álvaro Cunqueiro, Vigo (Pontevedra), España, 12Cardiología. Hospital Universitario Lucus Augusti, Lugo, España, 13Cardiología. Hospital Universitario de Burgos, Burgos, España y 14Cardiología. Hospital Clínico Universitario Virgen de la Arrixaca, Murcia, España.
Más comunicaciones de los autores
- Álvarez-García Revés, Reyes
- Bañuelos, Teresa
-
Bermejo Thomas, Javier
- 4016-7 - Gemelos digitales: nuevas estrategias personalizadas de ablación de fibrilación auricular
- 6105-3 - Canulación bilateral frente a unilateral en oxigenación con membrana extracorpórea veno-arterial periférica
- 4008-3 - Impacto de variantes sinónimas en las miocardiopatías hereditarias
- 5018-5 - Efecto del bloqueo completo de rama izquierda como factor determinante de pronóstico en pacientes intervenidos de reemplazo valvular aórtico transcatéter. Experiencia de un centro terciario
- 6031-195 - Datos en la vida real de cangrelor en procedimientos de cardiología intervencionista
- 6055-364 - El gradiente de presión intraventricular es el índice ecocardiográfico más sensible del estado inotrópico del ventrículo izquierdo
- 5020-6 - Implante de válvula aórtica transcatéter en paciente joven: experiencia y factores predictores de eventos en un hospital terciario
- 6030-178 - Predictores de recuperación de la función ventricular izquierda tras el implante de prótesis valvular aórtica percutánea
- 5018-2 - La esclerosis aórtica "funcional" se asocia a un aumento de riesgo de insuficiencia cardiaca y mortalidad
- 6114-9 - Perfil clínico e impacto pronóstico de la anemia en los pacientes sometidos a implante de válvula aórtica transcatéter
- 6123-16 - Malrotación de dispositivos de asistencia ventricular izquierda tipo Impella
- 5004-6 - Caracterización electrofisiológica del sustrato auricular mediante imagen electrocardiográfica (ECGi) sin necesidad de imagen cardiaca previa
- 5005-6 - Imagen de estasis mediante ecocardiografía para predecir el riesgo de ictus e infarto cerebral silente en los pacientes con miocardiopatía dilatada no isquémica
- 6110-9 - Estimación no invasiva de la presión capilar pulmonar mediante algoritmos de machine learning
- 4008-6 - ¿Es útil el screening familiar en miocardiopatía hipertrófica?
- 5010-2 - ¿Refleja el ensayo DanGer shock la vida real?
- 5009-6 - Disfunción endotelial microvascular y espasmo microvascular en pacientes con infarto agudo de miocardio con elevación del ST y enfermedad multivaso: prevalencia y predictores clínicos y angiográficos
- 5018-8 - Predictores de descompensación de insuficiencia cardiaca tras el implante de prótesis valvular aórtica percutánea
- Carrillo, Ana Paula
- Centeno Jiménez, Miriam
-
Espinosa Castro, M. Ángeles
- 4008-3 - Impacto de variantes sinónimas en las miocardiopatías hereditarias
- 4008-6 - ¿Es útil el screening familiar en miocardiopatía hipertrófica?
- 5005-6 - Imagen de estasis mediante ecocardiografía para predecir el riesgo de ictus e infarto cerebral silente en los pacientes con miocardiopatía dilatada no isquémica
- 5002-11 - Caracterización y pronóstico de la amiloidosis cardiaca hereditaria por transtirretina en España
- 5002-7 - Genotipos de alto riesgo en miocardiopatía dilatada: más allá de las arritmias
-
Fernández Ávila, Ana Isabel
- 4008-3 - Impacto de variantes sinónimas en las miocardiopatías hereditarias
- 5005-6 - Imagen de estasis mediante ecocardiografía para predecir el riesgo de ictus e infarto cerebral silente en los pacientes con miocardiopatía dilatada no isquémica
- 4008-6 - ¿Es útil el screening familiar en miocardiopatía hipertrófica?
- Garoux, Anaülle
- Gómez González, Cristina
- Medrano López, Constancio
- Méndez Fernández, Irene
-
Olsen Rodríguez, Renée
- 6104-3 - Implementación de una herramienta de inteligencia artificial en una consulta de cardiología general. Una nueva oportunidad
- 6101-4 - Concordancia entre intervenciones cardiológicas y propuestas de inteligencia artificial para pacientes que presentan dolor torácico, basadas en interconsultas desde el servicio de urgencias a cardiología
- 4009-4 - Implicaciones diagnósticas y pronosticas de los niveles de biomarcadores de inflamación y disfunción endotelial en el infarto de miocardio con y sin arterias coronarias obstructivas
- 4008-3 - Impacto de variantes sinónimas en las miocardiopatías hereditarias
- 6103-11 - MINOCA vs MICAD en el paciente anciano: características y pronóstico
- 6060-376 - Resonancia magnética cardiaca en pacientes con MINOCA en la práctica clínica habitual
- 6048-307 - Infarto de miocardio con arterias coronarias no obstructivas (MINOCA): características clínicas y pronóstico
- 6063-404 - Impacto de la edad en la insuficiencia cardiaca. Particularidades clínicas, epidemiológicas y pronósticas en el paciente anciano
- 6123-8 - Evolución a largo plazo y factores predictores en los pacientes con MINOCA
- 4008-6 - ¿Es útil el screening familiar en miocardiopatía hipertrófica?
- 6063-406 - Impacto de la hiponatremia en la hospitalización por insuficiencia cardiaca
- 6006-19 - Evaluación pronóstica de un grupo de pacientes con dolor torácico y curva plana de troponina
- 6010-46 - Diferencias entre hombres y mujeres menores de 65 años con infarto agudo de miocardio: una cohorte española
- 6048-306 - Perfil clínico y pronóstico de pacientes con infarto agudo de miocardio con arterias coronarias no obstruidas que hayan tenido un síndrome coronario agudo previo
- 6048-309 - El impacto de la enfermedad renal en pacientes con infarto agudo de miocardio con y sin lesiones obstructivas
- 6065-417 - Diferencias en cuanto al sexo en pacientes ingresados por insuficiencia cardiaca: características y pronóstico
- 6042-279 - Toma de decisiones en los pacientes con dolor torácico y curva plana de troponina
- 6048-308 - Implicaciones pronósticas de los pacientes diabéticos con MINOCA
- 5019-5 - Factores predictores de MINOCA en pacientes con infarto agudo de miocardio
- 6013-83 - Características clínicas y pronóstico del MINOCA en función de la edad
- 6045-295 - Infarto agudo de miocardio ¿ha cambiado el manejo en los últimos años?
- 6072-450 - Strain global longitudinal en insuficiencia cardiaca con fracción de eyección preservada. ¿Es útil?
- 6076-474 - Factores predictivos de ingreso hospitalario en pacientes con dolor torácico y curva plana de troponina
- Rojas, Alexis
- Vázquez Aguilera, Nélida
- Vilches Soria, Silvia