ISSN: 0300-8932
Factor de impacto 2024
4,9
SEC 2012 - El Congreso de las Enfermedades Cardiovasculares

Sevilla,
18 - 20 de Octubre de 2012
4042. Cardiopatía pediátrica-Cardiopatías congénitas I
Fecha
: 20-10-2012 00:00:00
Tipo
: Comunicaciones mini orales
Sala
: Sala C3 (Planta 1)
4042-7. Q447X. mutación sin sentido causal de una miocardiopatía izquierda dominante
Iván Gómez Milanés, Inmaculada Pérez Sánchez, Esperanza García-Molina, María Sabater Molina, David López Cuenca, Francisco Ruiz Espejo, Juan Ramón Gimeno Blanes y Mariano Valdés Chávarri del Servicio de Análisis Clínicos y Servicio de Cardiología del Hospital Universitario Virgen de la Arrixaca, Murcia.
Introducción: La miocardiopatía arritmogénica de ventrículo derecho (MAVD) es una enfermedad normalmente ocasionada por mutaciones en uno o más genes desmosómicos cardiacos. Algunas mutaciones en el gen de la desmoplaquina (DSP) dan lugar a un patrón de herencia autosómico dominante relacionado con la implicación del ventrículo izquierdo en la MAVD.
Métodos: Se realizó el estudio de una población de 28 pacientes compuesta por 14 mujeres de 38,5 ± 23,3 años y 14 varones de 36,5 ± 14,5 pertenecientes a 3 familias afectadas por MAVD, dos de ellas con historia familiar de muerte súbita. Esta cohorte se obtuvo de un screening de 64 pacientes con MAVD utilizando como técnica de cribado el dHPLC. El análisis de los 28 individuos se realizó mediante la secuenciación exónica y de las regiones intrónicas flanqueantes correspondientes al gen DSP utilizando el analizador ABI3130 y los softwares Seq Scape y Sequencing Analysis.
Resultados y conclusiones: Encontramos una variante génica (Q447X) de tipo "nonsense"(fig.) que no había sido descrita con anterioridad que da lugar a un patrón de herencia autosómico dominante con alta penetrancia (91%). Se produce una transición de C por T generándose un codón de stop que da lugar a un péptido un 85% menor que el wildtype. En la mayoría de los casos, las mutaciones originadas por un codón de stop son causa de la enfermedad. Once de los 28 pacientes estudiados eran portadores de la mutación, 10 de ellos afectados de MAVD. El resto eran pacientes sanos. De los 11 portadores, 8 eran mujeres con edades comprendidas entre los 21 y los 45 años y el resto varones con edades comprendidas entre los 16 y los 45 años. El aminoácido 447 se encuentra en uno de los dominios de la cabeza globular de la desmoplaquina que participa en la unión de esta proteína con placoglobina y placofilina. Es de destacar que varias mutaciones en este gen se han asociado con el desarrollo de miocardiopatía arritmogénica de predominio ventricular izquierdo e incluso con afectación aislada de este ventrículo y simulando una miocardiopatía dilatada idiopática. Concluimos que esta variante génica Q447X podría ser la causa de una MAVD con predominio en el ventrículo izquierdo.
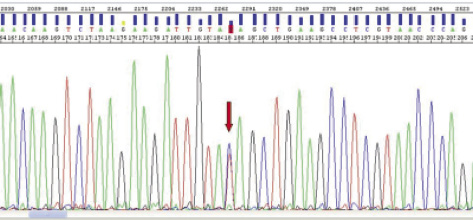
Mutación Q447X en el gen DSP.
Comunicaciones disponibles de "Cardiopatía pediátrica-Cardiopatías congénitas I"
- 4042-1. Presentación
- Carlos Brotons Cuixart, Barcelona y José Ignacio Carrasco Moreno, Valencia.
- 4042-2. Taquiarritmias auriculares tras cierre percutáneo de defectos grandes del tabique interauricular: incidencia y predictores
- Felipe Hernández Hernández, Rosana Hernández Antolín, José Antonio Baz Alonso, Federico Gimeno de Carlos, José M. de la Torre Hernández, Ángel Sánchez Recalde, Roberto Blanco Mata y José María Hernández García del Hospital 12 de Octubre, Madrid, Hospital Clínico San Carlos, Madrid y Hospital Meixoeiro, Vigo (Pontevedra).
- 4042-3. Reparación percutánea de la coartación aórtica compleja: resultados inmediatos y tardíos
- Elena Villanueva Fernández, José María Segura Saint-Gerons, Javier Suárez de Lezo Herreros de Tejada, Marta Santisteban Sánchez de Puerta, Miguel Romero Moreno, Djordje Pavlovic Djurovic, Manuel Pan Álvarez-Ossorio y José Suárez de Lezo Cruz Conde del Servicio de Cardiología del Hospital Reina Sofía, Córdoba.
- 4042-4. Miocardiopatía hipertrófica e implante de desfibrilador automático en la edad pediátrica
- Marta Ortega Molina, Jorge Figueroa, David Doiny, Sergio Castrejón-Castrejón, David Filgueiras-Rama, Luis García-Guereta, José Luis Merino y Federico Gutiérrez-Larraya de la Unidad de Arritmias y Electrofisiología Robotizada y Servicio de Cardiología Infantil del Hospital Universitario La Paz, Madrid.
- 4042-5. Patrones ecocardiográficos de síndrome de Shone incompleto en adultos con una coartación de aorta
- Luis González Torres, Rocío Gómez Domínguez, Blanca Muñoz Calero, Irene Méndez Santos, Pablo Bastos Amador, Luisa Cabeza Beltrán, María José Rodríguez Puras y Pastora Gallego García de Vinuesa del Área del Corazón del Hospital Universitario Virgen Macarena, Sevilla y Área del Corazón del Hospital Universitario Virgen del Rocío, Sevilla.
- 4042-6. La consulta de transición del paciente adolescente con cardiopatía congénita: experiencia de un centro de referencia nacional
- Alberto Núñez García, Enrique Maroto Álvaro, Raquel Prieto Arévalo, Fernando Sarnago Cebada, Constancio Medrano, María Ángeles Espinosa Castro, Pedro Luis Sánchez Fernández y Francisco Fernández-Avilés del Servicio de Cardiología y Servicio de Cardiología Pediátrica del Hospital General Universitario Gregorio Marañón, Madrid.
- 4042-7. Q447X. mutación sin sentido causal de una miocardiopatía izquierda dominante
- Iván Gómez Milanés, Inmaculada Pérez Sánchez, Esperanza García-Molina, María Sabater Molina, David López Cuenca, Francisco Ruiz Espejo, Juan Ramón Gimeno Blanes y Mariano Valdés Chávarri del Servicio de Análisis Clínicos y Servicio de Cardiología del Hospital Universitario Virgen de la Arrixaca, Murcia.
- 4042-8. Eplerenona en ventrículo derecho sistémico. ensayo clínico aleatorizado
- Laura Dos Subirana, Sandra Pujadas Olano, Montserrat Estruch Alrich, Assumpta Mas Mongay, Antònia Pijuan Doménech, Ricard Serra Grima, Maite Subirana Doménech y Jaume Casaldàliga-Ferrer del Hospital Universitario Vall d'Hebron, Barcelona y Hospital de la Santa Creu i Sant Pau, Barcelona.
- 4042-9. Conocimiento de la enfermedad y del pronóstico vital en adultos con cardiopatía congénita
- Blanca Muñoz Calero, Luis González Torres, María Rocío Gómez Domínguez, Irene Méndez Santos, Almudena Lloret Campoy, María José Rodríguez Puras, María Luisa Cabeza Letrán y Pastora Gallego García de Vinuesa del Área del Corazón (Cardiopatías Congénitas del Adulto) del Hospital Universitario Virgen Macarena y Hospital Universitario Virgen del Rocío, Sevilla.
- 4042-10. Síndrome de Williams-Beuren e intervalo QTc prolongado: ¿causa de muerte súbita cardiaca o hallazgo incidental?
- Diana Domingo Valero, José Ignacio Carrasco Moreno, Francisco Martínez Castellano, Miguel Ángel Arnau Vives, Joaquín Rueda Soriano, María Rodríguez Serrano, Antonio Salvador Sanz y Esther Zorio Grima del Hospital Universitario y Politécnico La Fe, Valencia.
- 4042-11. Conocimiento de la enfermedad y del riesgo de la gestación en mujeres con cardiopatía congénita en edad fértil
- Blanca Muñoz Calero, Luis González Torres, Irene Méndez Santos, María Rocío Gómez Domínguez, Pablo Bastos Amador, María José Rodríguez Puras, María Luisa Cabeza Letrán y Pastora Gallego García de Vinuesa del Área del Corazón (Cardiopatías Congénitas del Adulto) del Hospital Universitario Virgen Macarena y Hospital Universitario Virgen del Rocío, Sevilla.
- 4042-12. Estudio de la rentabilidad de los test genéticos en las cardiopatías hereditarias
- María Sabater Molina, Juan Ramón Gimeno Blanes, Esperanza García-Molina Sáez, Patricia Pascual Gilabert, Inmaculada Sánchez Pérez, Iván Gómez Milanés, Francisco Ruiz-Espejo y Mariano Valdés Chávarri del Servicio de Análisis Clínicos y Servicio de Cardiología del Hospital Universitario Virgen de la Arrixaca, Murcia.
Más comunicaciones de los autores
- García-Molina, Esperanza
-
Gimeno Blanes, Juan Ramón
- 4001-6 - Factores moduladores del grosor de la pared del ventrículo izquierdo en la miocardiopatía hipertrófica
- 6001-414 - R14Del, mutación holandesa del fosfolamban (PLN) en una familia española. Aspectos genotipo-fenotipo
- 6000-284 - Registro multicéntrico de dronedarona en miocardiopatía hipertrófica
- 4037-4 - Papel de la monitorización electrocardiográfica con Holter de 7 días en la detección de taquicardia ventricular no sostenida en pacientes con miocardiopatía hipertrófica
- 4037-3 - Efecto de la hipertensión arterial y el deporte en el grosor parietal ventricular en pacientes con miocardiopatía hipertrófica
- 6000-262 - Enfermedad de Anderson-Fabry: no tan "ligada a X"
- 4023-4 - Correlación genotipo-fenotipo de una nueva mutación causal para síndrome de QT largo tipo 2
- 4010-2 - Organización de un programa de screening cardiológico en deportistas. proyecto piloto
- 4001-5 - Influencia de la genética en el riesgo de muerte súbita en la miocardiopatía hipertrófica
- 6001-675 - Modificación de un stent coronario para realizar la técnica de Szabo
- 6000-287 - Penetrancia en relación a la edad en portadores genéticos de miocardiopatía hipertrófica
- 6000-286 - Complicaciones de dispositivos en pacientes con miocardiopatía y canalopatía tratados con marcapasos permanente y/o desfibriladores AUTOMÁTICOS implantables. Experiencia en nuestro centro
- 4042-12 - Estudio de la rentabilidad de los test genéticos en las cardiopatías hereditarias
- 4042-7 - Q447X. mutación sin sentido causal de una miocardiopatía izquierda dominante
-
Gómez Milanés, Iván
- 6000-287 - Penetrancia en relación a la edad en portadores genéticos de miocardiopatía hipertrófica
- 4042-7 - Q447X. mutación sin sentido causal de una miocardiopatía izquierda dominante
- 4042-12 - Estudio de la rentabilidad de los test genéticos en las cardiopatías hereditarias
- 6001-414 - R14Del, mutación holandesa del fosfolamban (PLN) en una familia española. Aspectos genotipo-fenotipo
- 4001-6 - Factores moduladores del grosor de la pared del ventrículo izquierdo en la miocardiopatía hipertrófica
-
López Cuenca, David
- 4037-4 - Papel de la monitorización electrocardiográfica con Holter de 7 días en la detección de taquicardia ventricular no sostenida en pacientes con miocardiopatía hipertrófica
- 6000-287 - Penetrancia en relación a la edad en portadores genéticos de miocardiopatía hipertrófica
- 6000-286 - Complicaciones de dispositivos en pacientes con miocardiopatía y canalopatía tratados con marcapasos permanente y/o desfibriladores AUTOMÁTICOS implantables. Experiencia en nuestro centro
- 4010-2 - Organización de un programa de screening cardiológico en deportistas. proyecto piloto
- 4042-7 - Q447X. mutación sin sentido causal de una miocardiopatía izquierda dominante
- 6000-262 - Enfermedad de Anderson-Fabry: no tan "ligada a X"
- 4001-6 - Factores moduladores del grosor de la pared del ventrículo izquierdo en la miocardiopatía hipertrófica
-
Pérez Sánchez, Inmaculada
- 4001-6 - Factores moduladores del grosor de la pared del ventrículo izquierdo en la miocardiopatía hipertrófica
- 6001-414 - R14Del, mutación holandesa del fosfolamban (PLN) en una familia española. Aspectos genotipo-fenotipo
- 4037-3 - Efecto de la hipertensión arterial y el deporte en el grosor parietal ventricular en pacientes con miocardiopatía hipertrófica
- 4042-7 - Q447X. mutación sin sentido causal de una miocardiopatía izquierda dominante
- 6000-287 - Penetrancia en relación a la edad en portadores genéticos de miocardiopatía hipertrófica
- 4037-4 - Papel de la monitorización electrocardiográfica con Holter de 7 días en la detección de taquicardia ventricular no sostenida en pacientes con miocardiopatía hipertrófica
-
Ruiz Espejo, Francisco
- 4001-5 - Influencia de la genética en el riesgo de muerte súbita en la miocardiopatía hipertrófica
- 6000-287 - Penetrancia en relación a la edad en portadores genéticos de miocardiopatía hipertrófica
- 4042-7 - Q447X. mutación sin sentido causal de una miocardiopatía izquierda dominante
- 4001-6 - Factores moduladores del grosor de la pared del ventrículo izquierdo en la miocardiopatía hipertrófica
- 6001-414 - R14Del, mutación holandesa del fosfolamban (PLN) en una familia española. Aspectos genotipo-fenotipo
-
Sabater Molina, María
- 6000-287 - Penetrancia en relación a la edad en portadores genéticos de miocardiopatía hipertrófica
- 4042-12 - Estudio de la rentabilidad de los test genéticos en las cardiopatías hereditarias
- 6001-414 - R14Del, mutación holandesa del fosfolamban (PLN) en una familia española. Aspectos genotipo-fenotipo
- 4001-6 - Factores moduladores del grosor de la pared del ventrículo izquierdo en la miocardiopatía hipertrófica
- 4042-7 - Q447X. mutación sin sentido causal de una miocardiopatía izquierda dominante
-
Valdés Chávarri, Mariano
- 6000-391 - Evolución temporal de los valores del NT-proBNP tras el trasplante cardiaco
- 6000-249 - Estenosis aórtica grave con bajos gradientes y función sistólica preservada. ¿una entidad real?
- 4042-12 - Estudio de la rentabilidad de los test genéticos en las cardiopatías hereditarias
- 4042-7 - Q447X. mutación sin sentido causal de una miocardiopatía izquierda dominante
- 6000-116 - Galectina-3 y remodelado cardiaco en un modelo animal de infarto de miocardio
- 6000-286 - Complicaciones de dispositivos en pacientes con miocardiopatía y canalopatía tratados con marcapasos permanente y/o desfibriladores AUTOMÁTICOS implantables. Experiencia en nuestro centro
- 4037-3 - Efecto de la hipertensión arterial y el deporte en el grosor parietal ventricular en pacientes con miocardiopatía hipertrófica
- 6000-118 - Predicción ecocardiográfica del tamaño del infarto de miocardio agudo y crónico en un modelo animal y evaluación del índice de funcionalidad miocárdico
- 6000-373 - Caracterización de las arritmias en la evolución del paciente trasplantado cardiaco
- 4028-2 - Los stents farmacoactivos han igualado el pronóstico de los diabéticos a los no diabéticos en el tratamiento de las oclusiones coronarias crónicas. Datos del estudio multicéntrico CIBELES
- 4021-7 - Metformina protege contra la cardiotoxicidad inducida por doxorrubicina a través de la super-regulación de la cadena pesada de la ferritina (FHC)
- 4023-4 - Correlación genotipo-fenotipo de una nueva mutación causal para síndrome de QT largo tipo 2
- 4021-5 - Identificación y confirmación de la haptoglobina como potencial biomarcador sérico en la miocardiopatía hipertrófica mediante técnicas de proteómica
- 4001-5 - Influencia de la genética en el riesgo de muerte súbita en la miocardiopatía hipertrófica
- 4010-2 - Organización de un programa de screening cardiológico en deportistas. proyecto piloto