ISSN: 0300-8932
Factor de impacto 2024
4,9
SEC 2013 - El Congreso de las Enfermedades Cardiovasculares

Valencia,
24 - 26 de Octubre de 2013
6015. Enfermedad miocardio/pericardio
Fecha
: 24-10-2013 18:32:00
Tipo
: Pósters
Sala
: PÓSTERS
6015-487. Mutación en cypher-zasp en pacientes con miocardiopatía arritmogénica de ventrículo derecho
José María López Ayala, Marina Navarro Peñalver, Iván Gómez Milanés, María José Oliva Sandoval, David López Cuenca, Josefa González Carrillo, Juan R. Gimeno Blanes y Mariano Valdés Chávarri del Hospital Universitario Virgen de la Arrixaca, Murcia.
Introducción: La miocardiopatía arritmogénica de ventrículo derecho (MAVD) es una enfermedad primaria del músculo cardiaco asociada a una elevada incidencia de muerte súbita y arritmias ventriculares. A pesar de estar originada por una disrupción de los desmosomas, en el 50% de los casos no se consigue identificar una mutación causal en un gen desmosómico. Mutaciones en otros genes no desmosómicos se han postulado como causa de la enfermedad.
Objetivos: Caracterización clínica y estudio genético en una familia de MAVD sin mutación causal identificada es genes desmosómicos
Métodos: Tras realizar el diagnostico de MAVD en el caso índice, se estudiaron 9 familiares. Se realizó una historia y exploración física completa, ECG, SAECG, ecocardiograma 2D-Doppler, RMN cardiaca, Holter de 24 horas y prueba de esfuerzo. Se extrajo una muestra de sangre periférica para estudio genético. Tras descartar mediante técnica de secuenciación convencional la presencia de una mutación causal en genes desmosómicos en el probando, se realizó estudio genético mediante Next Generation Sequencing. El diagnostico de MAVD se realizó según la Task Force Criteria de 2010.
Resultados: La mutación T351A en el gen LDB3 fue identificada en 6 familiares (mediana de edad 47 años; 4 mujeres). Mutaciones en el gen LDB3, que codifica la proteína sarcomérica Cypher-ZASP, se han descrito en casos de miocardiopatía dilatada y no compactación apical. 2 pacientes cumplieron criterios para el diagnostico definitivo de MAVD, 1 obtuvo diagnostico borderline y 3 fueron diagnosticados como posibles afectados. Ningún portador presentó signos de enfermedad neuromuscular. El caso índice debutó como muerte súbita recuperada y finalmente falleció como complicación del implante de un desfibrilador. 3 portadores presentaban una extensa afectación estructural de ventrículo derecho. En ningún portador se ha documentado arritmias ventriculares durante un seguimiento de 4 años.
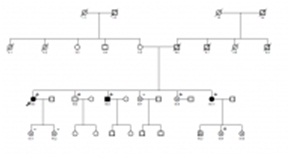
Figura. Árbol familiar.
Conclusiones: Por primera vez presentamos una familia con diagnóstico de MAVD portadora de una mutación en LDB3. Hasta la fecha, mutaciones en este gen se han asociado al desarrollo de miocardiopatía dilatada, miocardiopatía no compactada y distrofias musculares. Futuros estudios in vitro serán necesarios para determinar el mecanismo fisiopatológico por el que mutaciones en LDB3 pueden causar MAVD.
Comunicaciones disponibles de "Enfermedad miocardio/pericardio"
- 6015-474. La fibrilación auricular en la miocardiopatía dilatada afecta la función cardiopulmonar durante la realización del test de ergoespirometría
- Belén Marí-López1, Alberto Domínguez Rodríguez1, María del Carmen García-Baute1, Julia González1, Marta Padilla1, Esther González1, María Carrillo-Pérez Tomé1 y Pedro Abreu-González2 del 1Complejo Hospitalario Universitario de Canarias, San Cristóbal de La Laguna (Santa Cruz de Tenerife) y 2Universidad de La Laguna, San Cristóbal de La Laguna (Santa Cruz de Tenerife).
- 6015-475. Correlación entre parámetros electrocardiográficos y ecocardiográficos para el diagnóstico precoz de la enfermedad de Fabry
- Mónica Ramos Sánchez1, Caroline Coast2, Valentini Paresi2, Kalaiarasi Janagarajan2, Constantinos O'Mahony2 y Perry M. Elliott2 del 1Hospital Nuestra Señora de Sonsoles, Ávila y 2The Heart Hospital, London.
- 6015-476. Capacidad de ejercicio evaluada por test de ergoespirometría en los pacientes con miocardiopatía dilatada: efecto paradójico de la obesidad
- Julia González1, Alberto Domínguez Rodríguez1, María del Carmen García-Baute1, Belén Marí-López1, Marta Padilla1, Esther González1 y Pedro Abreu-González2 del 1Complejo Hospitalario Universitario de Canarias, San Cristóbal de La Laguna (Santa Cruz de Tenerife) y 2Universidad de La Laguna, San Cristóbal de La Laguna (Santa Cruz de Tenerife).
- 6015-477. Estudio sobre mutaciones somáticas en los genes MYH7, MYBPC3, TPM1, TNNT2 y TNNI3 en casos esporádicos de miocardiopatía hipertrófica
- Lucía Núñez1, Juan Ramón Gimeno-Blanes2, María Isabel Rodríguez-García1, Lorenzo Monserrat1, Esther Zorio3, Alfonso Castro-Beiras1 y Manuel Hermida Prieto4 del 1Instituto de Investigación Biomédica A Coruña-INIBIC, Servicio de Cardiología, CHUAC, UDC, A Coruña, 2Departamento de Cardiología, Hospital Universitario Virgen de la Arrixaca, Murcia, 3Departamento de Cardiología, Hospital Universitari i Politècnic La Fe, Valencia y 4Instituto de Investigación Biomédica A Coruña-INIBIC, Servicio de Cardiología, CHUAC,UDC, A Coruña.
- 6015-478. Importancia de la recuperación de la fracción de eyección y sus predictores en la miocardiopatía dilatada alcohólica
- Gonzalo Guzzo Merello, Pablo García-Pavía, Marta Cobo, Ana Briceño, Patricia Avellana, Manuel Gómez-Bueno, Javier Segovia y Luis Alonso-Pulpón del Hospital Universitario Puerta de Hierro, Majadahonda (Madrid).
- 6015-479. Tratamiento inicial del derrame pericárdico grave de origen tumoral mediante pericardiotomía percutánea con balón
- Juan Ruiz-García1, Santiago Jiménez-Valero2, Guillermo Galeote2, Ángel Sánchez-Recalde2, Sebastián Carrizo2, Nieves Montoro2, Ignacio Plaza3 y Raúl Moreno2 del 1Hospital Universitario La Paz, Madrid y Hospital Infanta Sofía, San Sebastián de los Reyes (Madrid), 2Hospital Universitario La Paz, Madrid y 3Hospital Infanta Sofía, San Sebastián de los Reyes (Madrid).
- 6015-480. Seguimiento a medio plazo de pacientes sintomáticos con miocardiopatía no compactada
- Joel Salazar-Mendiguchía1, José González-Costello1, Teresa Oliveras2, Francisco Gual2, Alejandro Ruiz Majoral1, Josep Lupón2, Nicolás Manito1 y Ángel Cequier1 del 1Área de Enfermedades del Corazón, Unidad de Miocardiopatías, Hospital Universitari de Bellvitge, L'Hospitalet de Llobregat (Barcelona) y 2Hospital Universitari Germans Trias i Pujol, Badalona (Barcelona).
- 6015-481. Perfil clínico de la miocarditis que precisa ingreso hospitalario
- Luis Mauricio Torres Sánchez, Marta Guillén Marzo, Nuria Farré López, Judit Rodríguez López y Alfredo Bardají Ruiz del Hospital Universitario Joan XXIII, Tarragona.
- 6015-482. Miopericarditis: Seguimiento a 50 meses
- Javier León Jiménez, Sergio Gamaza Chulián, María Recuerda Núñez, Santiago Jesús Camacho Freire, Alejandro Gutiérrez Barrios, Ana del Río Lechuga, Miguel Alba Sánchez y José Carlos Vargas-Machuca Caballero del Hospital del S.A.S. de Jerez de la Frontera, Cádiz.
- 6015-483. Manejo invasivo del derrame pericárdico (DP) de origen tumoral: características y resultados
- J. Goirigolzarri, E. Rodríguez, M.A. Restrepo, G. Guzzo, P. Martínez, L. Alonso-Pulpón, P. García Pavía y M. Cobo del Hospital Universitario Puerta de Hierro, Majadahonda (Madrid).
- 6015-484. Utilidad clínica de la biopsia endomiocárdica para diagnóstico de miocardiopatías
- Isabel Zegrí Reiriz, Juan Francisco Oteo Domínguez, Javier Segovia Cubero, José Ramón Domínguez Puente, Arturo García-Touchard, José Antonio Fernández-Díaz, Francisco Javier Goicolea Ruigómez y Luis Antonio Alonso-Pulpón del Hospital Universitario Puerta de Hierro, Madrid.
- 6015-485. Utilidad de las diferentes pruebas para el diagnóstico de la miocardiopatía arritmogénica asociada a muerte súbita familiar
- Begoña Igual Muñoz1, Ydelise Mercedes Rodríguez de Muñoz2, Alicia Maceira González1, Jordi Estornell Erill1, Pilar Molina Aguilar3, Juan Giner Blasco3, Diana Domingo Valero2 y Esther Zorio Grima2 del 1ERESA, Valencia, 2Hospital La Fe, Valencia y 3Instituto de Medicina Legal, Valencia.
- 6015-486. Función plaquetaria y síndrome de Takotsubo
- Iván Javier Núñez Gil, Esther Bernardo, Gisela Feltes Guzmán, David Vivas, Alberto de Agustín, Pilar Jiménez Quevedo, Carlos Macaya y Antonio Fernández Ortiz del Hospital Clínico San Carlos, Madrid.
- 6015-487. Mutación en cypher-zasp en pacientes con miocardiopatía arritmogénica de ventrículo derecho
- José María López Ayala, Marina Navarro Peñalver, Iván Gómez Milanés, María José Oliva Sandoval, David López Cuenca, Josefa González Carrillo, Juan R. Gimeno Blanes y Mariano Valdés Chávarri del Hospital Universitario Virgen de la Arrixaca, Murcia.
- 6015-488. Administración toracoscópica de células madre mesenquimales en el saco pericárdico para el tratamiento del infarto agudo de miocardio
- Belén Moreno Naranjo, Idoia Díaz-Güemes Martín-Portugués, Verónica Crisóstomo Ayala, Javier García Casado, Fei Sun, Juan Maestre Antequera, Claudia Báez Díaz y Francisco Miguel Sánchez Margallo del CCMI "Jesús", Cáceres.
- 6015-489. Predictores de mejoría a largo plazo de la clase funcional en pacientes con miocardiopatía hipertrófica obstructiva sometidos a ablación septal percutánea
- Luis Renier Gonçalves, Ignacio Jesús Amat Santos, Frank Sliwinski, Jairo Monedero, Héctor Cubero, Itziar Gómez, Federico Gimeno de Carlos y J. Alberto San Román Calvar del Hospital Clínico Universitario de Valladolid-Instituto de Ciencias del Corazón (ICICOR), Valladolid.
- 6015-490. Preexcitación electrocardiográfica en pacientes diagnosticados de cardiopatías familiares
- Mariela Salar Alcaraz1, Carmen Muñoz-Esparza2, Pablo Peñafiel Verdú1, José María López Ayala2, Marina Navarro Peñalver3, Mariano Valdés-Chávarri3, Arcadi García Alberola1 y Juan Ramón Gimeno Blanes2 de la 1Unidad de Electrofisiología, Hospital Clínico Universitario Virgen de la Arrixaca, El Palmar (Murcia), 2Unidad de Cardiopatías Hereditarias, Hospital Clínico Universitario Virgen de la Arrixaca, El Palmar (Murcia) y 3Hospital Clínico Universitario Virgen de la Arrixaca, El Palmar (Murcia).
- 6015-491. Taponamiento cardiaco: Causas y evolución
- Cristina Sánchez Enrique, Iván Núñez-Gil, Ana Viana, Jean Paul Vilchez Tschischke, Julián Palacios Rubio, Alberto Cecconi, Carlos Macaya y Antonio Fernández Ortiz del Hospital Clínico San Carlos, Madrid.
- 6015-492. Superposición entre canalopatías y miocardiopatías: TV catecolaminérgica y miocardiopatía no compactada
- Esperanza García-Molina Sáez, Carmen Muñoz-Esparza, Mariela Salar Alcaraz, Pablo Peñafiel-Verdú, J. Vicente Campos Peris, Josefa González-Carrillo, David López Cuenca y Juan R. Gimeno-Blanes del Hospital Clínico Universitario Virgen de la Arrixaca, El Palmar (Murcia).
- 6015-493. Efectividad en la mejora clínica de la miectomía ampliada en la miocardiopatía hipertrófica. ¿Justifica la relación riesgo-beneficio la indicación precoz?
- Tomás Heredia Cambra, Lucia Doñate Bertolin, Carlos Ernesto Hernández Acuña, Mona Schuler, Ana María Bel Mínguez, Manuel Pérez Guillén, Francisco José Valera Martínez y J. Anastasio Montero Argudo del Hospital Politécnico y Universitario La Fe, Valencia.
- 6015-494. Predictores clínicos de desarrollo de fibrosis miocárdica en la miocardiopatía hipertrófica: la importancia de la herencia
- Eduardo Zatarain Nicolás, Eduardo Villacorta Argüelles, Mª Ángeles Espinosa Castro, Raquel Yotti Álvarez, Esther Pérez David, Raquel Prieto Arévalo, Pedro Luis Sánchez Fernández y Francisco Fernández-Avilés del Hospital General Universitario Gregorio Marañón, Madrid.
- 6015-495. Global Function Index como marcador precoz de disfunción miocárdica en pacientes con enfermedad de Fabry
- Luis Miguel Rincón Díaz1, Covadonga Fernández-Golfín1, Viviana Serra1, J.A. Herrero2, J. Torras3, Eduardo Casas Rojo1, Miguel Castillo Orive1 y José Luis Zamorano Gómez1 del 1Hospital Universitario Ramón y Cajal, Madrid, 2Hospital Clínico San Carlos, Madrid y 3Hospital Universitari de Bellvitge, L'Hospitalet de Llobregat (Barcelona).
Más comunicaciones de los autores
-
Gimeno-Blanes, Juan Ramón
- 4029-8 - Aporte de la medida de troponina T ultrasensible en el seguimiento evolutivo de un grupo de riesgo de miocardiopatía hipertrófica
- 6019-625 - Perfil clínico de los síncopes en pacientes con patrón de Brugada
- 4011-5 - Importancia de los estudios bioinformáticos como herramienta complementaria en la clasificación de variantes en los genes desmosómicos
- 4012-5 - El electrocardiograma normal en miocardiopatía hipertrófica se asocia a un perfil clínico más benigno
- 4015-7 - Distribución de cardiopatías hereditarias en población inmigrante en una unidad de cardiopatías familiares en la región de Murcia
- 4019-6 - Evaluación de la precisión de las escalas de riesgo quirúrgicas euroscore II y STS en su aplicación al implante de prótesis aórtica percutánea
- 4008-3 - Miocardiopatía hipertrófica atípica ligada al cromosoma X por una mutación en el gen FLH1
- 4012-4 - Valoración del intervalo QT en bipedestación como una nueva herramienta diagnóstica para el síndrome de QT largo
- 4008-5 - Fenotipos mixtos en la valoración de cardiopatías hereditarias
- 4043-8 - Causas de muerte súbita en la región de Murcia (estudio PHEIDDIPIDES)
- 6005-236 - Cuantificación de troponina T ultrasensible en sujetos portadores no afectados de miocardiopatía hipertrófica
- 4043-11 - Polimorfismos en el sistema renina-angiotensina-aldosterona predicen la severidad de la hipertrofia del ventrículo izquierdo en la miocardiopatía hipertrófica
- 6005-226 - Nueva mutación en el gen MYBPC3 asociada a miocardiopatía hipertrófica
- 6002-147 - Estrategia farmacoinvasiva y angioplastia de rescate como tratamiento del infarto agudo de miocardio en hospitales comarcales de la región de Murcia
- 6015-477 - Estudio sobre mutaciones somáticas en los genes MYH7, MYBPC3, TPM1, TNNT2 y TNNI3 en casos esporádicos de miocardiopatía hipertrófica
- 6015-487 - Mutación en cypher-zasp en pacientes con miocardiopatía arritmogénica de ventrículo derecho
- 6009-363 - Insuficiencia renal aguda tras implante de prótesis aórtica percutánea: incidencia y recuperación intrahospitalaria
- 6015-490 - Preexcitación electrocardiográfica en pacientes diagnosticados de cardiopatías familiares
- 6015-492 - Superposición entre canalopatías y miocardiopatías: TV catecolaminérgica y miocardiopatía no compactada
- 4027-8 - Resultados de la angioplastia primaria en pacientes octogenarios y nonagenarios
-
Gómez Milanés, Iván
- 4029-8 - Aporte de la medida de troponina T ultrasensible en el seguimiento evolutivo de un grupo de riesgo de miocardiopatía hipertrófica
- 4011-5 - Importancia de los estudios bioinformáticos como herramienta complementaria en la clasificación de variantes en los genes desmosómicos
- 4043-11 - Polimorfismos en el sistema renina-angiotensina-aldosterona predicen la severidad de la hipertrofia del ventrículo izquierdo en la miocardiopatía hipertrófica
- 6005-226 - Nueva mutación en el gen MYBPC3 asociada a miocardiopatía hipertrófica
- 6015-487 - Mutación en cypher-zasp en pacientes con miocardiopatía arritmogénica de ventrículo derecho
-
González-Carrillo, Josefa
- 6007-287 - Inconsistencias de la clasificación de la estenosis aórtica en el cálculo del área valvular realizada por planimetría mediante ecocardiograma transesofágico tridimensional
- 6015-487 - Mutación en cypher-zasp en pacientes con miocardiopatía arritmogénica de ventrículo derecho
- 4003-7 - Fiabilidad de la valoración anatómica del área valvular aórtica por medio de ecocardiografía transesofágica tridimensional en tiempo real
- 6015-492 - Superposición entre canalopatías y miocardiopatías: TV catecolaminérgica y miocardiopatía no compactada
-
López Ayala, José María
- 6000-85 - Incidencia y gravedad de estenosis de venas pulmonares tras crioablación aislada y tras aislamiento mixto con criobalón y radiofrecuencia
- 4043-8 - Causas de muerte súbita en la región de Murcia (estudio PHEIDDIPIDES)
- 4008-3 - Miocardiopatía hipertrófica atípica ligada al cromosoma X por una mutación en el gen FLH1
- 4008-5 - Fenotipos mixtos en la valoración de cardiopatías hereditarias
- 6015-487 - Mutación en cypher-zasp en pacientes con miocardiopatía arritmogénica de ventrículo derecho
- 6015-490 - Preexcitación electrocardiográfica en pacientes diagnosticados de cardiopatías familiares
-
López Cuenca, David
- 4015-7 - Distribución de cardiopatías hereditarias en población inmigrante en una unidad de cardiopatías familiares en la región de Murcia
- 6015-492 - Superposición entre canalopatías y miocardiopatías: TV catecolaminérgica y miocardiopatía no compactada
- 6015-487 - Mutación en cypher-zasp en pacientes con miocardiopatía arritmogénica de ventrículo derecho
- 4043-11 - Polimorfismos en el sistema renina-angiotensina-aldosterona predicen la severidad de la hipertrofia del ventrículo izquierdo en la miocardiopatía hipertrófica
- 4043-8 - Causas de muerte súbita en la región de Murcia (estudio PHEIDDIPIDES)
- 6002-147 - Estrategia farmacoinvasiva y angioplastia de rescate como tratamiento del infarto agudo de miocardio en hospitales comarcales de la región de Murcia
- 4008-3 - Miocardiopatía hipertrófica atípica ligada al cromosoma X por una mutación en el gen FLH1
- 6005-226 - Nueva mutación en el gen MYBPC3 asociada a miocardiopatía hipertrófica
- 4011-5 - Importancia de los estudios bioinformáticos como herramienta complementaria en la clasificación de variantes en los genes desmosómicos
-
Navarro-Peñalver, Marina
- 6018-597 - Control de frecuencia cardiaca en insuficiencia cardiaca. Frecuencia basal frente a media en registros prolongados
- 4019-6 - Evaluación de la precisión de las escalas de riesgo quirúrgicas euroscore II y STS en su aplicación al implante de prótesis aórtica percutánea
- 4003-7 - Fiabilidad de la valoración anatómica del área valvular aórtica por medio de ecocardiografía transesofágica tridimensional en tiempo real
- 6015-487 - Mutación en cypher-zasp en pacientes con miocardiopatía arritmogénica de ventrículo derecho
- 4017-7 - Valor complementario del ancho de distribución eritrocitaria y la escala CRUSADE para predecir hemorragias en síndrome coronario agudo sin elevación del ST
- 6004-213 - Uso de estatinas a dosis elevadas en pacientes con síndrome coronario agudo: importancia de la diabetes mellitus
- 6015-490 - Preexcitación electrocardiográfica en pacientes diagnosticados de cardiopatías familiares
- 4003-3 - Consistencia de la nueva clasificación propuesta de estenosis aórtica severa: conocimientos derivados del ecocardiograma transesofágico 3D
- 4008-3 - Miocardiopatía hipertrófica atípica ligada al cromosoma X por una mutación en el gen FLH1
-
Oliva Sandoval, María José
- 6007-287 - Inconsistencias de la clasificación de la estenosis aórtica en el cálculo del área valvular realizada por planimetría mediante ecocardiograma transesofágico tridimensional
- 4003-3 - Consistencia de la nueva clasificación propuesta de estenosis aórtica severa: conocimientos derivados del ecocardiograma transesofágico 3D
- 6015-487 - Mutación en cypher-zasp en pacientes con miocardiopatía arritmogénica de ventrículo derecho
- 6005-226 - Nueva mutación en el gen MYBPC3 asociada a miocardiopatía hipertrófica
-
Valdés-Chávarri, Mariano
- 4003-3 - Consistencia de la nueva clasificación propuesta de estenosis aórtica severa: conocimientos derivados del ecocardiograma transesofágico 3D
- 6019-625 - Perfil clínico de los síncopes en pacientes con patrón de Brugada
- 6015-490 - Preexcitación electrocardiográfica en pacientes diagnosticados de cardiopatías familiares
- 6009-363 - Insuficiencia renal aguda tras implante de prótesis aórtica percutánea: incidencia y recuperación intrahospitalaria
- 6015-487 - Mutación en cypher-zasp en pacientes con miocardiopatía arritmogénica de ventrículo derecho
- 6018-584 - Micropartículas de pequeño tamaño en la insuficiencia cardiaca sistólica. Posible implicación fisiológica en la respuesta isquémica
- 6018-597 - Control de frecuencia cardiaca en insuficiencia cardiaca. Frecuencia basal frente a media en registros prolongados
- 4041-7 - Factores relacionados con la infrautilización de inhibidores del sistema renina-angiotensina en pacientes con síndrome coronario agudo
- 6002-152 - Fórmulas de filtrado glomerular CKD-EPI y escala de GRACE como predictores de mortalidad en pacientes con síndrome coronario agudo sin elevación del ST
- 4049-6 - ¿Podría tener la concentración de NT-proBNP un papel como predictor de ictus en pacientes con fibrilación auricular no valvular?
- 6005-240 - Fibrilación ventricular en cirugía cardiaca. Modelo de evolución natural de fibrilación en humanos
- 6005-239 - Modo de inicio de la fibrilación ventricular desde la asistolia
- 6004-213 - Uso de estatinas a dosis elevadas en pacientes con síndrome coronario agudo: importancia de la diabetes mellitus
- 4030-4 - Utilidad de un catéter de ablación con sensado de fuerza en la evaluación de las áreas de cicatriz miocárdicas en pacientes con taquicardia ventricular
- 4035-3 - La determinación de monómeros de fibrina aporta información pronóstica en los pacientes con fibrilación auricular no valvular bajo tratamiento anticoagulante
- 4008-5 - Fenotipos mixtos en la valoración de cardiopatías hereditarias
- 4012-4 - Valoración del intervalo QT en bipedestación como una nueva herramienta diagnóstica para el síndrome de QT largo
- 4017-7 - Valor complementario del ancho de distribución eritrocitaria y la escala CRUSADE para predecir hemorragias en síndrome coronario agudo sin elevación del ST
- 4003-4 - Insuficiencia aórtica residual tras implante de prótesis autoexpandibles. comparación entre prótesis aórticas percutáneas de Edwards y prótesis tipo Perceval
- 4019-6 - Evaluación de la precisión de las escalas de riesgo quirúrgicas euroscore II y STS en su aplicación al implante de prótesis aórtica percutánea
- 4015-7 - Distribución de cardiopatías hereditarias en población inmigrante en una unidad de cardiopatías familiares en la región de Murcia
- 4010-5 - Polimorfismo Val34Leu del Factor XIII: influencia en las características cinéticas y funcionales de trombos de sangre completa
- 4025-6 - ¿Debemos incluir la insuficiencia renal en las escalas de estimación del riesgo embólico en la fibrilación auricular no valvular?
- 4027-8 - Resultados de la angioplastia primaria en pacientes octogenarios y nonagenarios