ISSN: 0300-8932
Factor de impacto 2024
4,9
SEC 2021 - El Congreso de la Salud Cardiovascular

Zaragoza,
28 - 30 de Octubre de 2021
Introducción
Dr. Héctor Bueno
Presidente del Comité Científico del Congreso
Comité ejecutivo
Comité de evaluadores
Listado de sesiones
Índice de autores
4004. Hipertensión pulmonar en la práctica clínica
Fecha
: 28-10-2021 10:45:00
Tipo
: Comunicaciones orales
Sala
: Sala 23 (planta 2)
4004-2. VALOR PRONÓSTICO DE LAS VARIANTES GENÉTICAS EN LA HIPERTENSIÓN ARTERIAL PULMONAR. ¿SON PREOCUPANTES LAS VARIANTES EN BMPR2?
Alejandro Cruz Utrilla1, Natalia Gallego Gozalo2, María José Cristo Ropero1, Carmen Pérez-Olivares Delgado1, Jair Tenorio Castaño2, Pablo Lapunzina2, Manuel López Meseguer3, Amaya Martínez Meñaca4, Fernando Arribas Ynsaurriaga5 y M. Pilar Escribano Subias1
1Unidad de Hipertensión Pulmonar, Servicio de Cardiología, Hospital Universitario 12 de Octubre, Madrid. 2Instituto de Genética Médica y Molecular (INGEMM, Hospital Universitario La Paz, Madrid. 3Hospital Universitario Vall d'Hebron, Barcelona. 4Hospital Universitario Marqués de Valdecilla, Santander. 5Servicio de Cardiología, Hospital Universitario 12 de Octubre, Madrid.
1Unidad de Hipertensión Pulmonar, Servicio de Cardiología, Hospital Universitario 12 de Octubre, Madrid. 2Instituto de Genética Médica y Molecular (INGEMM, Hospital Universitario La Paz, Madrid. 3Hospital Universitario Vall d'Hebron, Barcelona. 4Hospital Universitario Marqués de Valdecilla, Santander. 5Servicio de Cardiología, Hospital Universitario 12 de Octubre, Madrid.
Introducción y objetivos: En los últimos años se han descubierto múltiples nuevas variantes genéticas implicadas en la fisiopatología de la hipertensión arterial pulmonar (HAP). Sin embargo, no se conoce el riesgo asociado al diagnóstico de estas mutaciones.
Métodos: Se incluyeron pacientes con el diagnóstico de HAP y con al menos un panel NGS de 21 genes realizado entre los años 2011-2020. Todos los pacientes firmaron consentimiento informado escrito. Se excluyó la enfermedad venooclusiva pulmonar, HAP asociada a cardiopatías congénitas o a enfermedades del tejido conectivo. Se utilizó test de Χ2 y ANOVA para la comparación de variables cualitativas y cuantitativas, respectivamente. Un análisis de supervivencia libre de muerte o trasplante pulmonar crudo y ajustado por edad entre los 3 grupos sirvió para evaluar el evento principal. Se utilizó un valor p < 0,05.
Resultados: Se incluyeron 361 pacientes. Las variantes patogénicas/probablemente patogénicas más habituales se localizaron en el gen BMPR2. Se detectaron además dos pacientes portadores de variantes en KCKN3, dos en ACVRL1 y un caso en los genes KCNA5, TBX4, CPS1 y GDF2. En comparación con el resto de los pacientes, los portadores de mutaciones presentaron una edad de diagnóstico menor y peores parámetros hemodinámicos. Estos pacientes alcanzaron mayores distancias en la prueba de la marcha de los 6 minutos (TM6M) realizado al diagnóstico. Los portadores de mutaciones en el gen BMPR2 también mostraron mayores valores de capacidad de difusión de monóxido de carbono (DLCO) en comparación con los otros dos grupos a estudio (tabla). Después de un seguimiento medio de 104 meses se apreció una tendencia no significativa en los pacientes portadores de mutaciones en BMPR2 hacia una mejor supervivencia, desapareciendo este hallazgo tras el ajuste por la edad al diagnóstico (fig.).
|
Características de los grupos de pacientes incluidos |
||||
|
No portadores (n = 323) |
BMPR2 (n = 33) |
Otras variantes genéticas (n = 8) |
Valor p |
|
|
Edad-años (media ± de) |
46,2 ± 0,9 |
35,9 ± 2,8 |
35,2 ± 5,6 |
< 0,001 |
|
Mujeres (n/%) |
218 (66,9) |
24 (72,7) |
5 (62,5) |
0,759 |
|
HAP familiar (n/%) |
8 (2,5) |
16 (48,5) |
1 (12,5) |
< 0,001 |
|
TAPSE-mm (media ± DE) |
17,3 ± 0,3 |
17,1 ± 1,1 |
16,2 ± 1,9 |
0,839 |
|
Área AD-mm2 (media ± DE) |
21,7 ± 0,7 |
20,0 ± 2,6 |
20,6 ± 5,4 |
0,806 |
|
IC-L/min/m2 (media ± DE) |
2,4 ± 0,1 |
2,1 ± 0,2 |
2,8 ± 0,3 |
0,806 |
|
PAPm-mmHg (media ± DE) |
52,0 ± 0,8 |
60,6 ± 2,4 |
66,0 ± 4,9 |
< 0,001 |
|
RVP-UW (media ± de) |
11,7 ± 0,3 |
16,1 ± 1,1 |
17,7 ± 2,2 |
< 0,001 |
|
DLCO -% (media ± DE) |
56,6 ± 1,4 |
80,5 ± 4,7 |
59,9 ± 10,0 |
< 0,001 |
|
Distancia TM6M-metros (media ± de) |
406,5 ± 7,0 |
448,4 ± 21,3 |
450,3 ± 42,6 |
0,116 |
|
Tratamiento con epoprostenol iv (n/%) |
71 (22,1) |
11 (33,3) |
3 (37,5) |
0,221 |
|
Estado al final de seguimiento (n/%) |
||||
|
Vivo |
219 (67,8) |
24 (72,7) |
5 (62,5) |
0,321 |
|
Muerto |
62 (19,2) |
2 (6,1) |
2 (25,0) |
|
|
Trasplante pulmonar |
42 (13,0) |
7 (21,2) |
1 (12,5) |
|
|
AD: aurícula derecha; IC: índice cardiaco; PAPm: presión arterial pulmonar media; RVP: resistencias vasculares pulmonares; DLCO: capacidad de difusión pulmonar para el monóxido de carbono; TM6M: test de la marcha 6 min. |
||||
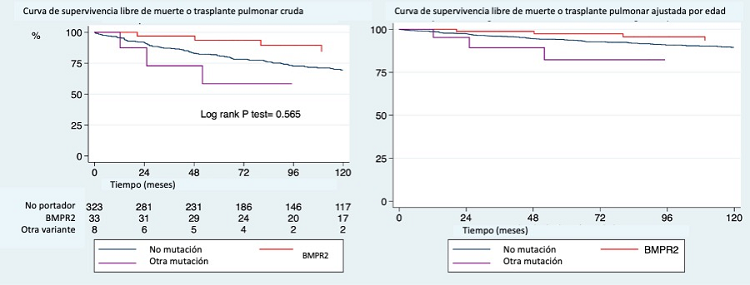
Conclusiones: Este trabajo recoge las variantes genéticas identificadas en una cohorte de pacientes con HAP idiopática o hereditaria en España. Las principales variantes identificadas fueron aquellas en BMPR2. En estos, a pesar de su mayor gravedad hemodinámica, su juventud, mejor capacidad funcional y la integridad de la membrana alveolocapilar podrían explicar el pronóstico comparable a otros casos de HAP idiopática. Sin embargo, los portadores de otras variantes genéticas presentaron una tendencia a un peor pronóstico a pesar de su juventud.
Comunicaciones disponibles de "Hipertensión pulmonar en la práctica clínica"
- 4004-1. MODERADOR
- Rafael Bravo Marqués, Málaga
- 4004-2. VALOR PRONÓSTICO DE LAS VARIANTES GENÉTICAS EN LA HIPERTENSIÓN ARTERIAL PULMONAR. ¿SON PREOCUPANTES LAS VARIANTES EN BMPR2?
- Alejandro Cruz Utrilla1, Natalia Gallego Gozalo2, María José Cristo Ropero1, Carmen Pérez-Olivares Delgado1, Jair Tenorio Castaño2, Pablo Lapunzina2, Manuel López Meseguer3, Amaya Martínez Meñaca4, Fernando Arribas Ynsaurriaga5 y M. Pilar Escribano Subias1
1Unidad de Hipertensión Pulmonar, Servicio de Cardiología, Hospital Universitario 12 de Octubre, Madrid. 2Instituto de Genética Médica y Molecular (INGEMM, Hospital Universitario La Paz, Madrid. 3Hospital Universitario Vall d'Hebron, Barcelona. 4Hospital Universitario Marqués de Valdecilla, Santander. 5Servicio de Cardiología, Hospital Universitario 12 de Octubre, Madrid.
- 4004-3. HIPERTENSIÓN ARTERIAL PULMONAR PEDIÁTRICA. ¿DEBEMOS CAMBIAR LAS ACTUALES RECOMENDACIONES DE ESTUDIO GENÉTICO?
- Alejandro Cruz Utrilla1, Natalia Gallego Gozazo2, Alba Torrent3, María Elvira Garrido-Lestache Rodríguez-Montes4, Inmaculada Guillén5, Sonia Arias6, Amparo Moya7, Alberto Mendoza8, Juana Espín9, María del Mar Rodríguez Vázquez10, Julia Playán Escribano11, Carlos Labrandero12, Jair Tenorio Castaño2, M. Pilar Escribano Subias1 y María Jesús del Cerro Marín4
1Unidad de Hipertensión Pulmonar, Servicio de Cardiología, Hospital Universitario 12 de Octubre, Madrid. 2Instituto de Genética Médica y Molecular (INGEMM), Hospital Universitario La Paz, Madrid. 3Servicio de Neumología Pediátrica, Hospital Universitario Vall d'Hebron, Barcelona. 4Servicio de Cardiología Pediátrica, Hospital Universitario Ramón y Cajal, Madrid. 5Servicio de Cardiología Pediátrica, Hospital Universitario Virgen del Rocío, Sevilla. 6Servicio de Cardiología Pediátrica, Hospital Universitario Infanta Cristina, Badajoz. 7Servicio de Cardiología Pediátrica, Hospital Universitario La Fe, Valencia. 8Servicio de Cardiología Pediátrica, Hospital Universitario 12 de Octubre, Madrid. 9Servicio de Cardiología Pediátrica, Hospital Clínico Universitario Virgen de la Arrixaca, Murcia. 10Servicio de Cardiología Pediátrica, Hospital Universitario Virgen de las Nieves, Granada. 11Servicio de Cardiología, Hospital Clínico San Carlos, Madrid. 12Servicio de Cardiología Pediátrica, Hospital Universitario La Paz, Madrid.
- 4004-4. HIPERTENSIÓN ARTERIAL PULMONAR ASOCIADA AL VIRUS DE LA INMUNODEFICIENCIA HUMANA EN ESPAÑA. EPIDEMIOLOGÍA, TRATAMIENTO Y PRONÓSTICO
- María Lázaro Salvador1, Clara Itziar Soto Abánades2, Manuel López-Meseguer3, Amaya Martínez Meñaca4, Gregorio Miguel Pérez Peñate5, Juan Antonio Domingo Morera6, Teresa Elías Hernández7, Raquel López Reyes8, Javier Segovia Cubero9, Ernest Sala Llinas10, Antonio Lara Padrón11, Pedro Bedate Díaz12, Águeda Aurtenetxe Pérez13, Isabel Blanco Vich14 y Pilar Escribano Subias15
1Servicio de Cardiología, Hospital Virgen de la Salud, Toledo. 2Servicio de Medicina Interna, Hospital Universitario La Paz, Madrid. 3Servicio de Neumología, Hospital Universitario Vall d´Hebron, Barcelona. 4Servicio de Neumología, Hospital Universitario Marqués de Valdecilla, Santander, Cantabria. 5Unidad de Circulación Pulmonar, Servicio de Neumología, Hospital Universitario de Gran Canaria Dr. Negrín, Las Palmas de Gran Canaria. 6Servicio de Neumología, Hospital Universitario Miguel Servet, Zaragoza. 7Unidad Médico-Quirúrgica de Enfermedades Respiratorias, Instituto de Biomedicina de Sevilla (IBiS), Hospital Universitario Virgen del Rocío, Sevilla y Centro de Investigación Biomédica en Red de Enfermedades Respiratorias (CIBERES), Madrid. 8Servicio de Neumología, Hospital Universitario y Politécnico La Fe, Valencia. 9Servicio de Cardiología, Hospital Universitario Puerta de Hierro-Majadahonda y Centro de Investigación Biomédica en Red de Enfermedades Cardiovasculares (CIBERCV), Instituto de Salud Carlos III, Madrid. 10Servicio de Neumología, Hospital Universitario Son Espases, Islas Baleares. 11Servicio de Cardiología, Hospital Universitario de Canarias, Santa Cruz de Tenerife. 12Servicio de Neumología, Hospital Universitario Central de Asturias, Oviedo. 13Servicio de Neumología, Hospital Universitario Basurto, Bilbao, Bizkaia. 14Servicio de Neumología, Unidad de Hipertensión Pulmonar, Hospital Clínic, Institut d'Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Barcelona y Centro de Investigación Biomédica en Red de Enfermedades Respiratorias (CIBERES), Madrid. 15Unidad de Hipertensión Pulmonar, Servicio de Cardiología, Hospital Universitario 12 de Octubre y Centro de Investigación Biomédica en Red de Enfermedades Cardiovasculares (CIBERCV), Instituto de Salud Carlos III, Madrid.
- 4004-5. VALOR AÑADIDO DE LA ERGOESPIROMETRÍA EN LA ESTIMACIÓN DEL RIESGO EN LA HIPERTENSIÓN ARTERIAL PULMONAR
- Raquel Luna López, María José Cristo Ropero, Alejandro Cruz Utrilla, Alicia Ruíz Martín, Teresa Segura de la Cal, Ángela Flox Camacho, Fernando Arribas Ynsaurriaga y Pilar Escribano Subias
Hospital Universitario 12 de Octubre, Madrid.
- 4004-6. EFICACIA, SEGURIDAD Y SUPERVIVENCIA DEL PRIMER PROGRAMA DE ANGIOPLASTIA PULMONAR EN PACIENTES CON HPTEC INOPERABLE EN UN CENTRO DE REFERENCIA NACIONAL. SIETE AÑOS DE EXPERIENCIA
- Mª Teresa Velázquez Martín, Agustín Albarrán González-Trevilla, Fernando Sarnago Cebada, Nicolás Maneiro Melón, Sergio Huertas Nieto, Allende Pilar Olazabal-Valiente, Alejandro Cruz Utrilla, Aníbal Ruiz Curiel, Macarena Otero Escudero, María Guisasola Cienfuegos, Alicia Ruiz Martín, María José Cristo Ropero, María Jesús López Gude, Yolanda Revilla Ostolaza y M. Pilar Escribano Subias
Hospital Universitario 12 de Octubre, Madrid.
- 4004-7. COMPLICACIONES ASOCIADAS A CATÉTERES CENTRALES DE INSERCIÓN PERIFÉRICA Y CATÉTERES TIPO HICKMAN EN PACIENTES CON HIPERTENSIÓN PULMONAR AVANZADA
- Williams Hinojosa Camargo1, Alba Cruz Galbán2, Alejandro Cruz Utrilla3, María José Cristo Ropero3, Francisco López Medrano3, Fernando Arribas Ynsaurriaga3, Itzíar Gómez1, Carolina Iglesias Echeverría1 y Pilar Escribano Subias3
1Instituto de Ciencias del Corazón (ICICOR), Valladolid. 2Complejo Asistencial Universitario de Salamanca. 3Hospital Universitario 12 de Octubre, Madrid.
Más comunicaciones de los autores
-
Arribas Ynsaurriaga, Fernando
- 6022-13 - PAPEL DE LA ENFERMERA EN LA PATOLOGÍA VALVULAR: FIGURA CENTRAL EN LA UNIDAD DE VALVULOPATÍAS
- 6038-2 - TÉCNICO ESPECIALISTA EN ECOCARDIOGRAFÍA: UNA FIGURA IMPRESCINDIBLE EN LA UNIDAD DE IMAGEN CARDIACA
- 6045-10 - MONITORIZACIÓN REMOTA AVANZADA: EXPERIENCIA PILOTO CON KARDIA 6L EN PACIENTES PORTADORES DE TERAPIAS DE RESINCRONIZACIÓN CARDIACA
- 5017-5 - VALORACIÓN DE LA CAPACIDAD FUNCIONAL EN HIPERTENSIÓN ARTERIAL PULMONAR ASOCIADA A ESCLERODERMIA. ROL DE LA ERGOESPIROMETRÍA
- 5026-8 - ESTUDIO ELECTROFISIOLÓGICO EN PACIENTES CON SOSPECHA DE TAQUICARDIA PAROXÍSTICA SUPRAVENTRICULAR NO DOCUMENTADA: EFICAZ Y SEGURO
- 6035-9 - ECOCARDIOGRAFÍA DE ESTRÉS CON REGADENOSÓN: UN CAMINO POR ANDAR
- 5020-2 - ANTICOAGULACIÓN EN LA FIBRILACIÓN AURICULAR DE PACIENTES MUY ANCIANOS CON RIVAROXABÁN
- 6042-5 - RIVAROXABÁN EN PACIENTES CON FIBRILACIÓN AURICULAR Y ARTERIOPATÍA PERIFÉRICA. DATOS DE UN ESTUDIO MULTICÉNTRICO
- 6021-4 - CATETERISMO DERECHO CON SOBRECARGA DE VOLUMEN EN EL DIAGNÓSTICO DIFERENCIAL Y MANEJO DE PACIENTES CON PERFIL CLÍNICO DE HIPERTENSIÓN PULMONAR ASOCIADA A CARDIOPATÍA IZQUIERDA EN UNA UNIDAD DE REFERENCIA
- 4004-7 - COMPLICACIONES ASOCIADAS A CATÉTERES CENTRALES DE INSERCIÓN PERIFÉRICA Y CATÉTERES TIPO HICKMAN EN PACIENTES CON HIPERTENSIÓN PULMONAR AVANZADA
- 6021-11 - AUMENTO DE LA SUPERVIVENCIA EN EL SÍNDROME DE EISENMENGER. ¿CÓMO SON LOS PACIENTES DE MAYOR EDAD?
- 6034-7 - IMPLICACIÓN CLÍNICA DE LAS COMPLICACIONES DETECTADAS EN PLANOS MODIFICADOS ADICIONALES A LOS ESTANDARIZADOS DE ECOCARDIOGRAFÍA TRANSTORÁCICA EN PACIENTES PORTADORES DE DISPOSITIVOS DE SOPORTE MECÁNICO Y CATÉTERES
- 6035-8 - ECOCARDIOGRAFÍA DE ESTRÉS CON REGADENOSÓN PARA LA VALORACIÓN DEL RIESGO CARDIOVASCULAR PERIOPERATORIO EN CIRUGÍA NO CARDIACA
- 6021-7 - UTILIDAD DEL ECOCARDIOGRAMA TRANSTORÁCICO PARA EL CRIBADO DE ANEURISMAS DE ARTERIA PULMONAR EN PACIENTES CON HIPERTENSIÓN ARTERIAL PULMONAR
- 6021-6 - ARTERIOGRAFÍA PULMONAR SELECTIVA EN LA HIPERTENSIÓN PULMONAR TROMBOEMBÓLICA CRÓNICA: CARACTERÍSTICAS TÉCNICAS Y SEGURIDAD EN LA SERIE DE UN CENTRO DE REFERENCIA
- 6048-8 - ABLACIÓN DEL NÓDULO AURÍCULOVENTRICULAR EN PACIENTES CON INSUFICIENCIA CARDIACA Y FIBRILACIÓN AURICULAR PERMANENTE: ¿CUÁNTO DEBEMOS ESPERAR?
- 6018-10 - PREDICTORES DE POSITIVIDAD EN ESTUDIO ELECTROFISIOLÓGICO Y DE RECIDIVA CLÍNICA EN PACIENTES CON SOSPECHA NO DOCUMENTADA DE TAQUICARDIA PAROXÍSTICA SUPRAVENTRICULAR
- 4001-7 - FORMACIÓN DE LA LESIÓN DE RADIOFRECUENCIA CONTROLADA POR TEMPERATURA EN TIEMPO REAL: LESIONES EX VIVO Y APLICACIÓN EN TAQUICARDIA AURICULAR MACRORREENTRANTE
- 6001-4 - VALIDACIÓN DE LA ESCALA 2MACE EN LA PREDICCIÓN DE EVENTOS CARDIOVASCULARES ADVERSOS EN PACIENTES CON FIBRILACIÓN AURICULAR ANTICOAGULADOS CON RIVAROXABÁN. ANÁLISIS DE LOS RESULTADOS DEL ESTUDIO NACIONAL EMIR
- 5005-3 - EXPERIENCIA INICIAL EN ESTIMULACIÓN DE RAMA IZQUIERDA CON CABLE DE HÉLICE RETRÁCTIL FRENTE A CABLE SIN LUMEN
- 6021-3 - ¿DEBEMOS ACTUALIZAR LA ESTRATIFICACIÓN DE RIESGO EN HIPERTENSIÓN ARTERIAL PULMONAR ASOCIADA A ESCLERODERMIA?
- 6021-10 - UTILIDAD DE LA ERGOESPIROMETRÍA PARA EL DIAGNÓSTICO Y LA MONITORIZACIÓN DE LA ENFERMEDAD VENOOCLUSIVA PULMONAR
- 6048-7 - ¿SE NECESITAN TÉCNICAS DE IMAGEN ADICIONALES PARA MINIMIZAR LA RADIACIÓN EN LA CRIOABLACIÓN DE FIBRILACIÓN AURICULAR? LA ESTRATEGIA VLADIMIR
- 6018-5 - LA ABLACIÓN EMPÍRICA DE VÍA LENTA EN PACIENTES CON TAQUICARDIA SUPRAVENTRICULAR PAROXÍSTICA SOSPECHADA PERO NO DOCUMENTADA NI INDUCIBLE ES EFICAZ EN LA REDUCCIÓN DE RECIDIVAS
- 4004-2 - VALOR PRONÓSTICO DE LAS VARIANTES GENÉTICAS EN LA HIPERTENSIÓN ARTERIAL PULMONAR. ¿SON PREOCUPANTES LAS VARIANTES EN BMPR2?
- 4004-5 - VALOR AÑADIDO DE LA ERGOESPIROMETRÍA EN LA ESTIMACIÓN DEL RIESGO EN LA HIPERTENSIÓN ARTERIAL PULMONAR
- 6029-10 - UTILIDAD DE PLANOS ECOCARDIOGRÁFICOS MODIFICADOS OFF-AXIS VIEWS FRENTE A CONVENCIONALES EN LA DETECCIÓN DE COMPLICACIONES EN PACIENTES PORTADORES DE DISPOSITIVOS DE SOPORTE MECÁNICO Y CATÉTERES CENTRALES
- 5012-4 - LA PRESENCIA DE ENFERMEDAD VASCULAR ES UN IMPORTANTE MARCADOR PRONÓSTICO EN PACIENTES CON FIBRILACIÓN AURICULAR. ANÁLISIS DEL ESTUDIO PROSPECTIVO Y MULTICÉNTRICO EMIR
-
Cristo Ropero, María José
- 4004-2 - VALOR PRONÓSTICO DE LAS VARIANTES GENÉTICAS EN LA HIPERTENSIÓN ARTERIAL PULMONAR. ¿SON PREOCUPANTES LAS VARIANTES EN BMPR2?
- 4004-7 - COMPLICACIONES ASOCIADAS A CATÉTERES CENTRALES DE INSERCIÓN PERIFÉRICA Y CATÉTERES TIPO HICKMAN EN PACIENTES CON HIPERTENSIÓN PULMONAR AVANZADA
- 4004-5 - VALOR AÑADIDO DE LA ERGOESPIROMETRÍA EN LA ESTIMACIÓN DEL RIESGO EN LA HIPERTENSIÓN ARTERIAL PULMONAR
- 6021-11 - AUMENTO DE LA SUPERVIVENCIA EN EL SÍNDROME DE EISENMENGER. ¿CÓMO SON LOS PACIENTES DE MAYOR EDAD?
- 6021-2 - COVID-19 EN LOS PACIENTES CON HIPERTENSIÓN PULMONAR. EXPERIENCIA DE UN CENTRO
- 6021-10 - UTILIDAD DE LA ERGOESPIROMETRÍA PARA EL DIAGNÓSTICO Y LA MONITORIZACIÓN DE LA ENFERMEDAD VENOOCLUSIVA PULMONAR
- 6021-3 - ¿DEBEMOS ACTUALIZAR LA ESTRATIFICACIÓN DE RIESGO EN HIPERTENSIÓN ARTERIAL PULMONAR ASOCIADA A ESCLERODERMIA?
- 5017-5 - VALORACIÓN DE LA CAPACIDAD FUNCIONAL EN HIPERTENSIÓN ARTERIAL PULMONAR ASOCIADA A ESCLERODERMIA. ROL DE LA ERGOESPIROMETRÍA
- 4004-6 - EFICACIA, SEGURIDAD Y SUPERVIVENCIA DEL PRIMER PROGRAMA DE ANGIOPLASTIA PULMONAR EN PACIENTES CON HPTEC INOPERABLE EN UN CENTRO DE REFERENCIA NACIONAL. SIETE AÑOS DE EXPERIENCIA
-
Cruz Utrilla, Alejandro
- 4004-3 - HIPERTENSIÓN ARTERIAL PULMONAR PEDIÁTRICA. ¿DEBEMOS CAMBIAR LAS ACTUALES RECOMENDACIONES DE ESTUDIO GENÉTICO?
- 6021-4 - CATETERISMO DERECHO CON SOBRECARGA DE VOLUMEN EN EL DIAGNÓSTICO DIFERENCIAL Y MANEJO DE PACIENTES CON PERFIL CLÍNICO DE HIPERTENSIÓN PULMONAR ASOCIADA A CARDIOPATÍA IZQUIERDA EN UNA UNIDAD DE REFERENCIA
- 6031-7 - NUEVAS VÍAS GENÉTICO-MOLECULARES EN LA HIPERTENSIÓN ARTERIAL PULMONAR ASOCIADA A ENFERMEDADES DEL TEJIDO CONECTIVO
- 5017-5 - VALORACIÓN DE LA CAPACIDAD FUNCIONAL EN HIPERTENSIÓN ARTERIAL PULMONAR ASOCIADA A ESCLERODERMIA. ROL DE LA ERGOESPIROMETRÍA
- 4004-5 - VALOR AÑADIDO DE LA ERGOESPIROMETRÍA EN LA ESTIMACIÓN DEL RIESGO EN LA HIPERTENSIÓN ARTERIAL PULMONAR
- 4004-2 - VALOR PRONÓSTICO DE LAS VARIANTES GENÉTICAS EN LA HIPERTENSIÓN ARTERIAL PULMONAR. ¿SON PREOCUPANTES LAS VARIANTES EN BMPR2?
- 4004-6 - EFICACIA, SEGURIDAD Y SUPERVIVENCIA DEL PRIMER PROGRAMA DE ANGIOPLASTIA PULMONAR EN PACIENTES CON HPTEC INOPERABLE EN UN CENTRO DE REFERENCIA NACIONAL. SIETE AÑOS DE EXPERIENCIA
- 6021-10 - UTILIDAD DE LA ERGOESPIROMETRÍA PARA EL DIAGNÓSTICO Y LA MONITORIZACIÓN DE LA ENFERMEDAD VENOOCLUSIVA PULMONAR
- 6021-3 - ¿DEBEMOS ACTUALIZAR LA ESTRATIFICACIÓN DE RIESGO EN HIPERTENSIÓN ARTERIAL PULMONAR ASOCIADA A ESCLERODERMIA?
- 6021-2 - COVID-19 EN LOS PACIENTES CON HIPERTENSIÓN PULMONAR. EXPERIENCIA DE UN CENTRO
- 6021-11 - AUMENTO DE LA SUPERVIVENCIA EN EL SÍNDROME DE EISENMENGER. ¿CÓMO SON LOS PACIENTES DE MAYOR EDAD?
- 4004-7 - COMPLICACIONES ASOCIADAS A CATÉTERES CENTRALES DE INSERCIÓN PERIFÉRICA Y CATÉTERES TIPO HICKMAN EN PACIENTES CON HIPERTENSIÓN PULMONAR AVANZADA
-
Escribano Subias, M. Pilar
- 4004-6 - EFICACIA, SEGURIDAD Y SUPERVIVENCIA DEL PRIMER PROGRAMA DE ANGIOPLASTIA PULMONAR EN PACIENTES CON HPTEC INOPERABLE EN UN CENTRO DE REFERENCIA NACIONAL. SIETE AÑOS DE EXPERIENCIA
- 6021-3 - ¿DEBEMOS ACTUALIZAR LA ESTRATIFICACIÓN DE RIESGO EN HIPERTENSIÓN ARTERIAL PULMONAR ASOCIADA A ESCLERODERMIA?
- 4004-4 - HIPERTENSIÓN ARTERIAL PULMONAR ASOCIADA AL VIRUS DE LA INMUNODEFICIENCIA HUMANA EN ESPAÑA. EPIDEMIOLOGÍA, TRATAMIENTO Y PRONÓSTICO
- 6021-8 - EL PACIENTE CON SÍNDROME DE DOWN E HIPERTENSIÓN ARTERIAL PULMONAR ASOCIADA A CARDIOPATÍAS CONGÉNITAS: DE LA INFANCIA A LA EDAD ADULTA
- 5017-5 - VALORACIÓN DE LA CAPACIDAD FUNCIONAL EN HIPERTENSIÓN ARTERIAL PULMONAR ASOCIADA A ESCLERODERMIA. ROL DE LA ERGOESPIROMETRÍA
- 6021-10 - UTILIDAD DE LA ERGOESPIROMETRÍA PARA EL DIAGNÓSTICO Y LA MONITORIZACIÓN DE LA ENFERMEDAD VENOOCLUSIVA PULMONAR
- 6021-11 - AUMENTO DE LA SUPERVIVENCIA EN EL SÍNDROME DE EISENMENGER. ¿CÓMO SON LOS PACIENTES DE MAYOR EDAD?
- 4004-3 - HIPERTENSIÓN ARTERIAL PULMONAR PEDIÁTRICA. ¿DEBEMOS CAMBIAR LAS ACTUALES RECOMENDACIONES DE ESTUDIO GENÉTICO?
- 6021-2 - COVID-19 EN LOS PACIENTES CON HIPERTENSIÓN PULMONAR. EXPERIENCIA DE UN CENTRO
- 6021-7 - UTILIDAD DEL ECOCARDIOGRAMA TRANSTORÁCICO PARA EL CRIBADO DE ANEURISMAS DE ARTERIA PULMONAR EN PACIENTES CON HIPERTENSIÓN ARTERIAL PULMONAR
- 6021-6 - ARTERIOGRAFÍA PULMONAR SELECTIVA EN LA HIPERTENSIÓN PULMONAR TROMBOEMBÓLICA CRÓNICA: CARACTERÍSTICAS TÉCNICAS Y SEGURIDAD EN LA SERIE DE UN CENTRO DE REFERENCIA
- 6021-4 - CATETERISMO DERECHO CON SOBRECARGA DE VOLUMEN EN EL DIAGNÓSTICO DIFERENCIAL Y MANEJO DE PACIENTES CON PERFIL CLÍNICO DE HIPERTENSIÓN PULMONAR ASOCIADA A CARDIOPATÍA IZQUIERDA EN UNA UNIDAD DE REFERENCIA
- 4004-7 - COMPLICACIONES ASOCIADAS A CATÉTERES CENTRALES DE INSERCIÓN PERIFÉRICA Y CATÉTERES TIPO HICKMAN EN PACIENTES CON HIPERTENSIÓN PULMONAR AVANZADA
- 4004-5 - VALOR AÑADIDO DE LA ERGOESPIROMETRÍA EN LA ESTIMACIÓN DEL RIESGO EN LA HIPERTENSIÓN ARTERIAL PULMONAR
- 6031-7 - NUEVAS VÍAS GENÉTICO-MOLECULARES EN LA HIPERTENSIÓN ARTERIAL PULMONAR ASOCIADA A ENFERMEDADES DEL TEJIDO CONECTIVO
- 4004-2 - VALOR PRONÓSTICO DE LAS VARIANTES GENÉTICAS EN LA HIPERTENSIÓN ARTERIAL PULMONAR. ¿SON PREOCUPANTES LAS VARIANTES EN BMPR2?
-
Gallego Gozalo, Natalia
- 4004-3 - HIPERTENSIÓN ARTERIAL PULMONAR PEDIÁTRICA. ¿DEBEMOS CAMBIAR LAS ACTUALES RECOMENDACIONES DE ESTUDIO GENÉTICO?
- 4004-2 - VALOR PRONÓSTICO DE LAS VARIANTES GENÉTICAS EN LA HIPERTENSIÓN ARTERIAL PULMONAR. ¿SON PREOCUPANTES LAS VARIANTES EN BMPR2?
- 6031-7 - NUEVAS VÍAS GENÉTICO-MOLECULARES EN LA HIPERTENSIÓN ARTERIAL PULMONAR ASOCIADA A ENFERMEDADES DEL TEJIDO CONECTIVO
- Lapunzina, Pablo
- López Meseguer, Manuel
-
Martínez Meñaca, Amaya
- 4004-2 - VALOR PRONÓSTICO DE LAS VARIANTES GENÉTICAS EN LA HIPERTENSIÓN ARTERIAL PULMONAR. ¿SON PREOCUPANTES LAS VARIANTES EN BMPR2?
- 6021-8 - EL PACIENTE CON SÍNDROME DE DOWN E HIPERTENSIÓN ARTERIAL PULMONAR ASOCIADA A CARDIOPATÍAS CONGÉNITAS: DE LA INFANCIA A LA EDAD ADULTA
- 4004-4 - HIPERTENSIÓN ARTERIAL PULMONAR ASOCIADA AL VIRUS DE LA INMUNODEFICIENCIA HUMANA EN ESPAÑA. EPIDEMIOLOGÍA, TRATAMIENTO Y PRONÓSTICO
-
Pérez-Olivares Delgado, Carmen
- 4004-2 - VALOR PRONÓSTICO DE LAS VARIANTES GENÉTICAS EN LA HIPERTENSIÓN ARTERIAL PULMONAR. ¿SON PREOCUPANTES LAS VARIANTES EN BMPR2?
- 6021-10 - UTILIDAD DE LA ERGOESPIROMETRÍA PARA EL DIAGNÓSTICO Y LA MONITORIZACIÓN DE LA ENFERMEDAD VENOOCLUSIVA PULMONAR
- 6021-3 - ¿DEBEMOS ACTUALIZAR LA ESTRATIFICACIÓN DE RIESGO EN HIPERTENSIÓN ARTERIAL PULMONAR ASOCIADA A ESCLERODERMIA?
- 6021-2 - COVID-19 EN LOS PACIENTES CON HIPERTENSIÓN PULMONAR. EXPERIENCIA DE UN CENTRO
- 5017-5 - VALORACIÓN DE LA CAPACIDAD FUNCIONAL EN HIPERTENSIÓN ARTERIAL PULMONAR ASOCIADA A ESCLERODERMIA. ROL DE LA ERGOESPIROMETRÍA
- 6031-7 - NUEVAS VÍAS GENÉTICO-MOLECULARES EN LA HIPERTENSIÓN ARTERIAL PULMONAR ASOCIADA A ENFERMEDADES DEL TEJIDO CONECTIVO
-
Tenorio Castaño, Jair
- 4004-3 - HIPERTENSIÓN ARTERIAL PULMONAR PEDIÁTRICA. ¿DEBEMOS CAMBIAR LAS ACTUALES RECOMENDACIONES DE ESTUDIO GENÉTICO?
- 4004-2 - VALOR PRONÓSTICO DE LAS VARIANTES GENÉTICAS EN LA HIPERTENSIÓN ARTERIAL PULMONAR. ¿SON PREOCUPANTES LAS VARIANTES EN BMPR2?
- 6031-7 - NUEVAS VÍAS GENÉTICO-MOLECULARES EN LA HIPERTENSIÓN ARTERIAL PULMONAR ASOCIADA A ENFERMEDADES DEL TEJIDO CONECTIVO