ISSN: 0300-8932
Factor de impacto 2024
4,9
SEC 2021 - El Congreso de la Salud Cardiovascular

Zaragoza,
28 - 30 de Octubre de 2021
Introducción
Dr. Héctor Bueno
Presidente del Comité Científico del Congreso
Comité ejecutivo
Comité de evaluadores
Listado de sesiones
Índice de autores
5029. Cardiopatías familiares y genética cardiovascular II
Fecha
: 30-10-2021 15:00:00
Tipo
: Comunicaciones mini orales
Sala
: Sala Hiberus 2 (Hotel Hiberus)
5029-2. HISTORIA NATURAL DE LA MIOCARDIOPATÍA DILATADA POR VARIANTES EN MYH7
Fernando de Frutos Seminario1, Fernando Domínguez1, Juan Pablo Ochoa1, Juan Ramón Gimeno Blanes2, Folkert W. Asselbergs3, Torsten B. Rasmussen4, Esther Zorio Grima5, Mª Ángeles Espinosa Castro6, Rebeca Lorca Gutiérrez7, Job A. J. Verdonschot8, Pablo Elpidio García Granja9, Zofia T. Biliska10, Mª Eugenia Fuentes Cañamero11, José Manuel García Pinilla12 y Pablo García Pavía1
1Hospital Universitario Puerta de Hierro, Majadahonda, Madrid. 2Hospital Clínico Universitario Virgen de la Arrixaca, Murcia. 3Universitair Medisch Centrum Utrecht (Países Bajos). 4Aarhus Universitetshospital, Aarhus (Dinamarca). 5Hospital Universitario La Fe, Valencia. 6Hospital General Universitario Gregorio Marañón, Madrid. 7Hospital Universitario Central de Asturias, Oviedo, Asrturias. 8Maastricht Universitair Medisch Centrum, Maastricht (Países Bajos). 9Hospital Clínico Universitario de Valladolid. 10Uniwersytet Kardynala Stefana Wyszynskiegow, Varsovia (Polonia). 11Complejo Hospitalario Universitario de Badajoz. 12Hospital Clínico Universitario Virgen de la Victoria, Málaga.
1Hospital Universitario Puerta de Hierro, Majadahonda, Madrid. 2Hospital Clínico Universitario Virgen de la Arrixaca, Murcia. 3Universitair Medisch Centrum Utrecht (Países Bajos). 4Aarhus Universitetshospital, Aarhus (Dinamarca). 5Hospital Universitario La Fe, Valencia. 6Hospital General Universitario Gregorio Marañón, Madrid. 7Hospital Universitario Central de Asturias, Oviedo, Asrturias. 8Maastricht Universitair Medisch Centrum, Maastricht (Países Bajos). 9Hospital Clínico Universitario de Valladolid. 10Uniwersytet Kardynala Stefana Wyszynskiegow, Varsovia (Polonia). 11Complejo Hospitalario Universitario de Badajoz. 12Hospital Clínico Universitario Virgen de la Victoria, Málaga.
Introducción y objetivos: Las variantes en MYH7 son responsables de entre el 1-5% de los casos de miocardiopatía dilatada no isquémica (MCD). La información acerca de su curso clínico es escasa y está basada en pequeñas series. Buscamos describir la historia natural de la MCD por variantes en MYH7 así como establecer la relación entre la localización de las variantes y la expresión fenotípica.
Métodos: Se realizó un registro retrospectivo internacional de sujetos portadores de variantes patogénicas o posiblemente patogénicas en MYH7 asociadas al desarrollo de MCD. Se recogieron las características clínicas, electrocardiográficas y de imagen, así como los eventos arrítmicos (descarga de DAI, taquicardia ventricular o muerte súbita) o de insuficiencia cardiaca (IC) avanzada (implantación de asistencia ventricular, trasplante cardiaco o muerte por IC) durante el seguimiento. Los pacientes se agruparon en 4 categorías en función de la localización proteica de las variantes: S1, Región conversora, S2 y LMM.
Resultados: Se incluyeron 177 pacientes (edad media 39 ± 19,6 años, 57% varones, 73% con MCD en la evaluación inicial) procedentes de 32 centros con un tiempo mediano de seguimiento de 4,2 años (RIQ: 1,7-7,7). La tabla muestra las principales características clínicas de los pacientes. La edad media al diagnóstico de MCD fue 39,8 ± 19,5 años con una penetrancia del 84% a los 60 años. 20 pacientes (14%) se diagnosticaron con 10% o FEVI final > 50% con un incremento absoluto > 5%) durante el seguimiento. La tasa de eventos arrítmicos fue baja (3,1% a 5 años) y ocurrió en todos los casos en pacientes con FEVI ≤ 35%. Un total de 12 pacientes (8,6%) recibieron un trasplante cardiaco y 8 (5,8%) fallecieron por IC. Los pacientes con variantes en la región conversora presentaron un mayor riesgo de eventos arrítmicos y de IC (HR 7,98, IC95%: 3,2-19,8; p < 0,001).
|
Características clínicas |
||||
|
Global (N = 177) |
Sin MCD en primera evaluación (N = 47) |
MCD en primera evaluación (N = 130) |
p |
|
|
Edad (años) |
38,8 (19,6) |
31,3 (19,6) |
41,5 (19,0) |
0,003 |
|
Sexo femenino |
77 (43,5%) |
25 (55,3%) |
51 (39,2%) |
0,06 |
|
Enfermedad neuromuscular |
2 (1,1%) |
1 (2,1%) |
1 (0,8%) |
0,45 |
|
Fibrilación auricular previa |
11 (7,3%) |
0 (0%) |
11 (9,7%) |
0,046 |
|
Dispositivos |
0,32 |
|||
|
Marcapasos |
3 (1,7%) |
0 (0%) |
3 (2,3%) |
|
|
DAI |
3 (1,7%) |
0 (0%) |
3 (2,3%) |
|
|
Morfología del QRS |
0,35 |
|||
|
BRD |
7 (4,2%) |
1 (2,4%) |
6 (4,8%) |
|
|
BRI |
12 (7,3%) |
1 (2,4%) |
11 (8,9%) |
|
|
TICV |
18 (10,9%) |
3 (7,3%) |
15 (12,1%) |
|
|
Extrasístoles ventriculares (> 500/día) |
8 (10,4%) |
1 (5,6%) |
7 (11,9%) |
0,44 |
|
TVNS |
16 (16,8%) |
1 (4,6%) |
14 (20,5%) |
0,08 |
|
Grosor miocárdico, mm |
9,6 (2,0) |
9,1 (2,0) |
9,8 (1,9) |
0,10 |
|
FEVI (%) |
41,1 (14,9) |
58,9 (4,8) |
34,6 (11,7) |
< 0,001 |
|
No compactación |
60 (34,5%) |
12 (25,5%) |
48 (37,8%) |
0,13 |
|
TAPSE (< 17 mm) |
8 (9,9%) |
0 (0%) |
8 (12,5%) |
0,13 |
|
RTG en RM |
20 (27%) |
0 (0%) |
20 (33,9%) |
0,008 |
|
BRD: bloqueo de rama derecha; BRI: bloqueo de rama izquierda; TICV: trastorno inespecífico de la conducción ventricular; FEVI: fracción de eyección del ventrículo izquierdo; RTG: realce tardío gadolinio. |
||||
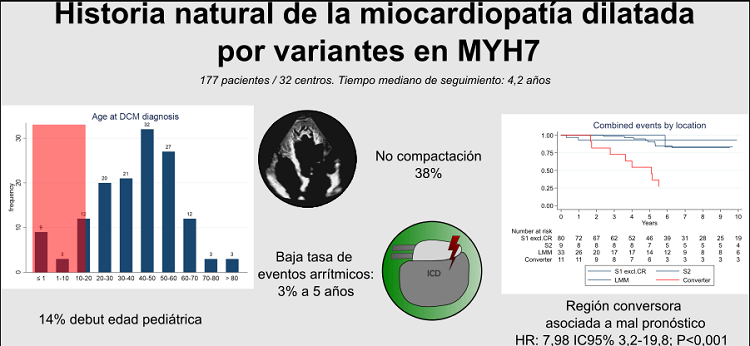
Figura central.
Conclusiones: La MCD por variantes en MYH7 se caracteriza por presentar una elevada tasa de diagnósticos a edades tempranas, una tasa de remodelado inverso baja, y rasgo de no compactación en un número significativo de pacientes. La incidencia de eventos arrítmicos y de IC avanzada es menor que en otras causas genéticas de MCD y las variantes en la región conversora se asocian a un peor pronóstico.
Comunicaciones disponibles de "Cardiopatías familiares y genética cardiovascular II"
- 5029-1. MODERADOR
- Carles Díez López, Barcelona
- 5029-2. HISTORIA NATURAL DE LA MIOCARDIOPATÍA DILATADA POR VARIANTES EN MYH7
- Fernando de Frutos Seminario1, Fernando Domínguez1, Juan Pablo Ochoa1, Juan Ramón Gimeno Blanes2, Folkert W. Asselbergs3, Torsten B. Rasmussen4, Esther Zorio Grima5, Mª Ángeles Espinosa Castro6, Rebeca Lorca Gutiérrez7, Job A. J. Verdonschot8, Pablo Elpidio García Granja9, Zofia T. Biliska10, Mª Eugenia Fuentes Cañamero11, José Manuel García Pinilla12 y Pablo García Pavía1
1Hospital Universitario Puerta de Hierro, Majadahonda, Madrid. 2Hospital Clínico Universitario Virgen de la Arrixaca, Murcia. 3Universitair Medisch Centrum Utrecht (Países Bajos). 4Aarhus Universitetshospital, Aarhus (Dinamarca). 5Hospital Universitario La Fe, Valencia. 6Hospital General Universitario Gregorio Marañón, Madrid. 7Hospital Universitario Central de Asturias, Oviedo, Asrturias. 8Maastricht Universitair Medisch Centrum, Maastricht (Países Bajos). 9Hospital Clínico Universitario de Valladolid. 10Uniwersytet Kardynala Stefana Wyszynskiegow, Varsovia (Polonia). 11Complejo Hospitalario Universitario de Badajoz. 12Hospital Clínico Universitario Virgen de la Victoria, Málaga.
- 5029-3. VARIANTES MISSENSE EN TTN COMO CAUSA DE MIOCARDIOPATÍA DILATADA
- Fernando Domínguez Rodríguez1, Laura Lalaguna2, Elías Herrero Galán2, Jesús Piqueras Flores3, Giovanna Giovinazzo1, Juan Pablo Ochoa1, Luis Enrique Escobar López1, Fernando de Frutos Seminario1, Esther González López1, Jorge Alegre Cebollada2, Enrique Lara Pezzi2 y Pablo García Pavía1
1Hospital Universitario Puerta de Hierro, Majadahonda, Madrid. 2Centro Nacional de Investigaciones Cardiovasculares, Madrid. 3Hospital General Universitario de Ciudad Real.
- 5029-4. ¿ES LA AFECTACIÓN DE VENTRÍCULO DERECHO UN FACTOR PRONÓSTICO EN EL PACIENTE CON MIOCARDIOPATÍA HIPERTRÓFICA?
- Milena Antúnez Ballesteros, Eduardo Villacorta Argüelles, Manuel Barreiro Pérez, Olga Cabañas Tendero, Mónica García Monsalvo, Fabián Blanco Fernández, Ana Elvira Laffond, Jean Carlos Núñez, Javier González Martín, Ángel Hernández Martos, Miguel Hernández Hidalgo, Lucía Rodríguez Estévez, Víctor Eduardo Vallejo García, Elena Díaz Peláez y Pedro Luis Sánchez Fernández
Complejo Asistencial Universitario de Salamanca.
- 5029-5. LA GAMMAGRAFÍA CON DPD NO DETECTA LA AFECTACIÓN CARDIACA POR AMILOIDOSIS TRANSTIRRETINA EN ENFERMEDAD DE INICIO PRECOZ ATTRV30M
- Aleix Olivella San Emeterio1, Sergi Yun1, Yassin Belahnech2, Carles Díez-López1, Laura García-Sánchez1, Carlos Casasnovas1, Emma González-Vilatarsana1, Laura Lladó1, Carme Baliellas1, José Rodríguez-Palomares2, Javier Limeres Freire2, Josep Comín Colet1 y José González Costello1
1Hospital de Bellvitge, L'Hospitalet de Llobregat, Barcelona. 2Hospital Universitario Vall d'Hebron, Barcelona.
- 5029-6. DISYUNCIÓN DEL ANILLO MITRAL EN PACIENTES CON AORTOPATÍAS HEREDITARIAS SINDRÓMICAS
- María Luz Servato, Ángela López-Sainz, Filipa Valente, Rubén Fernández-Galera, Guillem Casas-Masnou, Laura Gutiérrez García-Moreno, Javier Limeres Freire, Laura Galian Gay, Augusto Sao Aviles, M. Teresa González Alujas, José Fernando Rodríguez Palomares, Arturo Evangelista Masip y Gisela Teixido-Tura
Hospital Universitari Vall d'Hebron, Barcelona.
- 5029-7. RED ESPAÑOLA DE PATOLOGÍA AÓRTICA GENÉTICA (REPAG). UNA HERRAMIENTA PARA AVANZAR EN EL CONOCIMIENTO Y MANEJO DE UNA PATOLOGÍA EMERGENTE
- Gisela Teixido-Tura1, Alberto Forteza Gil2, Fernando Cabrera Bueno3, Julián Palomino-Doza4, Francisco Calvo Iglesias5, J. Francisco Nistal Herrera6, Amparo Hernándiz Martínez7, Rocío García Orta8, Rosario Sánchez Martínez9, Roberto Barriales Villa10, Elena Díaz Peláez11, Elena Montañés Delmas12, Francesca Perín13, Anna Sabaté Rotés14 y Arturo Evangelista Masip1
1Servicio de Cardiología, Hospital Universitari Vall d'Hebron, CIBERCV, Barcelona. 2Servicio de Cirugía Cardiaca, Hospital Puerta de Hierro, Madrid. 3Hospital Universitario Virgen de la Victoria, IBIMA, UMA, CIBERCV, Málaga. 4Servicio de Cardiología, Hospital Universitario 12 de Octubre, Madrid. 5Servicio de Cardiología, Hospital Universitario Álvaro Cunqueiro, Vigo, Pontevedra. 6Hospital Universitario Marqués de Valdecilla, IDIVAL, Universidad de Cantabria, Santander, Cantabria. 7IIS Hospital Universitario La Fe, Valencia. 8Servicio de Cardiología, Hospital Universitario Virgen de las Nieves, Granada. 9Hospital General Universitario de Alicante. 10Unidad de Cardiopatías Familiares, Complexo Hospitalario Universitario A Coruña. 11Complejo Asistencial Universitario de Salamanca, IBSAL, CIBERCV. 12Cardiología Pediátrica, Hospital Universitario 12 de Octubre, Madrid. 13Cardiología Pediátrica, Hospital Universitario Virgen de las Nieves, Granada. 14Cardiología Pediátrica, Hospital Universitari Vall d'Hebron, Barcelona.
- 5029-8. ESTUDIO GENÉTICO Y CRIBADO FAMILIAR EN PATOLOGÍA AÓRTICA FAMILIAR
- Maeve Soto Pérez1, Martín Negreira Caamaño1, Jesús Piqueras Flores2, Pedro Pérez Díaz1, Arancha González Marín3, Ignacio Sánchez Pérez4, Francisco Javier Jiménez Díaz5, José María Arizón Muñoz1, Álvaro Moreno Reig1, Manuel Rayo Gutiérrez1, Daniel Salas Bravo1, Jorge Martínez del Río1, Manuel Muñoz García1, Cristina Mateo Gómez1 y Andrez Felipe Cubides Novoa1
1Servicio de Cardiología, Hospital General Universitario de Ciudad Real. 2Unidad de Cardiopatías Familiares, Servicio de Cardiología, Hospital General Universitario de Ciudad Real. 3Sección de Cardiología Pediátrica, Servicio de Pediatría, Hospital General Universitario de Ciudad Real. 4Sección de Hemodinámica, Servicio de Cardiología, Hospital General Universitario de Ciudad Real. 5Sección de Arritmias y Electrofisiología, Servicio de Cardiología, Hospital General Universitario de Ciudad Real.
Más comunicaciones de los autores
- Asselbergs, Folkert W.
- Biliska, Zofia T.
- de Frutos Seminario, Fernando
- Domínguez Benito, Fernando
-
Espinosa Castro, M. Ángeles
- 4016-4 - COMBINACIÓN DE REALCE TARDÍO DE GADOLINIO Y GENOTIPO PARA EVALUAR EL PRONÓSTICO EN LA MIOCARDIOPATÍA DILATADA NO ISQUÉMICA
- 4016-3 - IMPACTO PRONÓSTICO DEL GENOTIPO EN LA MIOCARDIOPATÍA DILATADA NO ISQUÉMICA
- 6035-9 - ECOCARDIOGRAFÍA DE ESTRÉS CON REGADENOSÓN: UN CAMINO POR ANDAR
- 4016-2 - SCORE PREDICTIVO DE UN RESULTADO GENÉTICO POSITIVO EN LA MIOCARDIOPATÍA DILATADA IDIOPÁTICA
- 6020-10 - ANÁLISIS DE LAS COMPLICACIONES CARDIOVASCULARES DURANTE LA GESTACIÓN EN MUJERES CON MIOCARDIOPATÍA SEGUIDAS EN UN HOSPITAL TERCIARIO
- 5029-2 - HISTORIA NATURAL DE LA MIOCARDIOPATÍA DILATADA POR VARIANTES EN MYH7
- Fuentes Cañamero, M. Eugenia
-
García Granja, Pablo Elpidio
- 5019-3 - PREDICTORES DE MORTALIDAD HOSPITALARIA EN ENDOCARDITIS INFECCIOSA SOBRE VÁLVULAS PROTÉSICAS
- 5029-2 - HISTORIA NATURAL DE LA MIOCARDIOPATÍA DILATADA POR VARIANTES EN MYH7
- 6010-2 - ENDOCARDITIS INFECCIOSA POR CANDIDA ALBICANS Y CANDIDA PARAPSILOSIS: UN ESTUDIO COMPARATIVO
- 5019-2 - INDICADORES PRONÓSTICOS DE MORTALIDAD HOSPITALARIA EN LA ENDOCARDITIS INFECCIOSA SOBRE VÁLVULA NATIVA IZQUIERDA
- 6010-8 - ¿LOS FACTORES PRONÓSTICOS DE LA ENDOCARDITIS INFECCIOSA NATIVA SON IGUALES QUE LOS DE LA ENDOCARDITIS PROTÉSICA?
-
García Pavía, Pablo
- 6040-10 - BIOPSIA ENDOMIOCÁRDICA EN PACIENTES CON SOSPECHA DE MIOCARDIOPATÍA INFILTRATIVA
- 4016-4 - COMBINACIÓN DE REALCE TARDÍO DE GADOLINIO Y GENOTIPO PARA EVALUAR EL PRONÓSTICO EN LA MIOCARDIOPATÍA DILATADA NO ISQUÉMICA
- 4016-2 - SCORE PREDICTIVO DE UN RESULTADO GENÉTICO POSITIVO EN LA MIOCARDIOPATÍA DILATADA IDIOPÁTICA
- 4016-3 - IMPACTO PRONÓSTICO DEL GENOTIPO EN LA MIOCARDIOPATÍA DILATADA NO ISQUÉMICA
- 4016-7 - PRONÓSTICO DE PACIENTES CON NO COMPACTACIÓN DEL VENTRÍCULO IZQUIERDO Y FUNCIÓN SISTÓLICA CONSERVADA
- 5029-2 - HISTORIA NATURAL DE LA MIOCARDIOPATÍA DILATADA POR VARIANTES EN MYH7
- 5029-3 - VARIANTES MISSENSE EN TTN COMO CAUSA DE MIOCARDIOPATÍA DILATADA
-
García Pinilla, José Manuel
- 4016-2 - SCORE PREDICTIVO DE UN RESULTADO GENÉTICO POSITIVO EN LA MIOCARDIOPATÍA DILATADA IDIOPÁTICA
- 6031-10 - INCIDENCIA Y UTILIDAD DE LA DISFUNCIÓN VENTRICULAR DERECHA EN LA SELECCIÓN DE IMPLANTE DE DESFIBRILADOR EN LOS PORTADORES DE MUTACIONES DESMOSÓMICAS
- 4025-7 - ESTUDIO MULTICÉNTRICO ESPAÑOL SOBRE LA MONITORIZACIÓN E INCIDENCIA DE HIPERPOTASEMIA EN PACIENTES CON INSUFICIENCIA CARDIACA Y FRACCIÓN EYECCIÓN REDUCIDA. SPANIK-HF
- 4025-6 - SPANIK-HF-ESTUDIO MULTICÉNTRICO ESPAÑOL SOBRE LA PREVALENCIA, INCIDENCIA Y PRONÓSTICO DE HIPERPOTASEMIA EN PACIENTES CON INSUFICIENCIA CARDIACA: DATOS BASALES
- 5029-2 - HISTORIA NATURAL DE LA MIOCARDIOPATÍA DILATADA POR VARIANTES EN MYH7
- 6033-2 - SITUACIÓN DE LAS UNIDADES CARDIORRENALES EN ESPAÑA
- 6027-14 - PERSISTENCIA DEL TRATAMIENTO CON RIVAROXABÁN EN PACIENTES CON INSUFICIENCIA CARDIACA Y FIBRILACIÓN AURICULAR. DATOS DEL ESTUDIO FARAONIC
- 4016-7 - PRONÓSTICO DE PACIENTES CON NO COMPACTACIÓN DEL VENTRÍCULO IZQUIERDO Y FUNCIÓN SISTÓLICA CONSERVADA
- 4016-3 - IMPACTO PRONÓSTICO DEL GENOTIPO EN LA MIOCARDIOPATÍA DILATADA NO ISQUÉMICA
- 4016-4 - COMBINACIÓN DE REALCE TARDÍO DE GADOLINIO Y GENOTIPO PARA EVALUAR EL PRONÓSTICO EN LA MIOCARDIOPATÍA DILATADA NO ISQUÉMICA
-
Gimeno Blanes, Juan Ramón
- 4016-6 - LA VÍA EP300/TP53 SE ENCUENTRA ACTIVADA MIENTRAS QUE WNT E HIPPO SUPRIMIDAS EN MIOCARDIO DE PACIENTES CON MIOCARDIOPATÍA ARRITMOGÉNICA TAMBIÉN SIN INSUFICIENCIA CARDIACA
- 4016-2 - SCORE PREDICTIVO DE UN RESULTADO GENÉTICO POSITIVO EN LA MIOCARDIOPATÍA DILATADA IDIOPÁTICA
- 6031-15 - IMPACTO DE LA PREMATURIDAD EN EL DESARROLLO DE MIOCARDIOPATÍAS DE ORIGEN GENÉTICO EN LA EDAD PEDIÁTRICA
- 4016-4 - COMBINACIÓN DE REALCE TARDÍO DE GADOLINIO Y GENOTIPO PARA EVALUAR EL PRONÓSTICO EN LA MIOCARDIOPATÍA DILATADA NO ISQUÉMICA
- 4016-3 - IMPACTO PRONÓSTICO DEL GENOTIPO EN LA MIOCARDIOPATÍA DILATADA NO ISQUÉMICA
- 5029-2 - HISTORIA NATURAL DE LA MIOCARDIOPATÍA DILATADA POR VARIANTES EN MYH7
- 4016-7 - PRONÓSTICO DE PACIENTES CON NO COMPACTACIÓN DEL VENTRÍCULO IZQUIERDO Y FUNCIÓN SISTÓLICA CONSERVADA
- 6040-5 - RELACIÓN DEL NIVEL DE ACTIVIDAD FÍSICA Y EL CURSO EVOLUTIVO DE LA ENFERMEDAD EN PACIENTES CON SÍNDROME DE BRUGADA
-
Lorca Gutiérrez, Rebeca
- 5029-2 - HISTORIA NATURAL DE LA MIOCARDIOPATÍA DILATADA POR VARIANTES EN MYH7
- 5008-7 - ASOCIACIÓN DEL POLIMORFISMO RS2075744 EN EL ARN LARGO NO CODIFICANTE H19 CON EL RIESGO DE DESARROLLAR MIOCARDIOPATÍA HIPERTRÓFICA
- 6040-14 - EVOLUCIÓN A LARGO PLAZO DE PACIENTES CON MIOCARDIOPATÍA HIPERTRÓFICA PORTADORES DE DESFIBRILADOR AUTOMÁTICO IMPLANTABLE EN PREVENCIÓN PRIMARIA Y SECUNDARIA
-
Ochoa, Juan Pablo
- 4016-2 - SCORE PREDICTIVO DE UN RESULTADO GENÉTICO POSITIVO EN LA MIOCARDIOPATÍA DILATADA IDIOPÁTICA
- 5029-2 - HISTORIA NATURAL DE LA MIOCARDIOPATÍA DILATADA POR VARIANTES EN MYH7
- 6040-8 - LA GENÉTICA COMO AYUDA A LA ENTELEQUIA DE LA NO COMPACTACIÓN MIOCÁRDICA
- 5029-3 - VARIANTES MISSENSE EN TTN COMO CAUSA DE MIOCARDIOPATÍA DILATADA
- 6014-16 - VARIANTES GENÉTICAS COMO DETERMINANTES DEL PRONÓSTICO EN LA MIOCARDIOPATÍA ASOCIADA A LAMINA A/C
- 4016-3 - IMPACTO PRONÓSTICO DEL GENOTIPO EN LA MIOCARDIOPATÍA DILATADA NO ISQUÉMICA
- 4016-4 - COMBINACIÓN DE REALCE TARDÍO DE GADOLINIO Y GENOTIPO PARA EVALUAR EL PRONÓSTICO EN LA MIOCARDIOPATÍA DILATADA NO ISQUÉMICA
- Rasmussen, Torsten B.
- Verdonschot, Job A. J.
-
Zorio Grima, Esther
- 6040-9 - NUEVA FUENTE DE CULTIVOS PARA EL ESTUDIO DE CARDIOPATÍAS A PARTIR DE MUESTRAS FORENSES
- 6040-17 - PERFIL CLÍNICO-EPIDEMIOLÓGICO DE LAS ANOMALÍAS CORONARIAS COMO CAUSA DE MUERTE SÚBITA
- 5029-2 - HISTORIA NATURAL DE LA MIOCARDIOPATÍA DILATADA POR VARIANTES EN MYH7
- 6031-5 - POTENCIAL AYUDA DIAGNÓSTICA DE LA INMUNOFLUORESCENCIA DE DESMOPLAQUINA EN QUERATINOCITOS BUCALES PARA FAMILIAS CON MIOCARDIOPATÍA ARRITMOGÉNICA
- 5008-4 - EN BUSCA DE TEJIDOS SUBROGADOS PARA EL MIOCARDIO: LA METILACIÓN DEL PROMOTOR DEL GEN DE DESMOPLAQUINA EN QUERATINOCITOS BUCALES CORRELACIONA CON LA DEL MIOCARDIO
- 4016-7 - PRONÓSTICO DE PACIENTES CON NO COMPACTACIÓN DEL VENTRÍCULO IZQUIERDO Y FUNCIÓN SISTÓLICA CONSERVADA
- 6031-3 - ARTERIAS CORONARIAS CON NACIMIENTO EN SENO CONTRALATERAL ¿QUÉ PERFIL CLÍNICO-EPIDEMIOLÓGICO PRESENTAN EN UNA SERIE FORENSE DE MUERTE SÚBITA?
- 5008-2 - LAS VÍAS WNT, HIPPO Y ADIPOGÉNESIS, TAMBIÉN ALTERADAS EN GRASA EPICÁRDICA DE PACIENTES CON MIOCARDIOPATÍA ARRITMOGÉNICA A TRAVÉS DE LA DISREGULACIÓN DE MIRNA
- 4016-6 - LA VÍA EP300/TP53 SE ENCUENTRA ACTIVADA MIENTRAS QUE WNT E HIPPO SUPRIMIDAS EN MIOCARDIO DE PACIENTES CON MIOCARDIOPATÍA ARRITMOGÉNICA TAMBIÉN SIN INSUFICIENCIA CARDIACA