ISSN: 0300-8932
Factor de impacto 2024
4,9
SEC 2022 - El Congreso de la Salud Cardiovascular

Palma de Mallorca y online,
20 - 22 de Octubre de 2022
Introducción
Dr. Juan José Gómez Doblas
Presidente del Comité Científico del Congreso
Comité ejecutivo
Comité de evaluadores
Listado de sesiones
Índice de autores
4019. Registros uni y multicéntricos en cardiopatías familiares
Fecha
: 21-10-2022 15:30:00
Tipo
: Comunicaciones orales
Sala
: Sala Menorca 1 (Planta 3)
4019-5. RELEVANCIA DE LAS VARIANTES EN EL GEN OBSCN QUE GENERAN TRUNCAMIENTOS EN PACIENTES CON MIOCARDIOPATÍAS
María del Carmen García del Rey1, Ana I. Fernández Ávila2, María Ángeles Espinosa1, Silvia Vilches Soria1, Cristina Gómez González1, Irene Méndez Fernández1, Javier Bermejo Thomas1 y Francisco Fernández-Avilés1
1Hospital General Universitario Gregorio Marañón, Madrid y 2CIBERCV. Instituto de Investigación Sanitaria Gregorio Marañón, Madrid.
1Hospital General Universitario Gregorio Marañón, Madrid y 2CIBERCV. Instituto de Investigación Sanitaria Gregorio Marañón, Madrid.
Introducción y objetivos: Las oscurinas son proteínas sarcoméricas codificadas por el gen OBSCN con funciones estructurales y reguladoras en estrecha relación con titina. Estudios recientes han relacionado las variantes que causan haploinsuficiencia en OBSCN con miocardiopatías, incluyendo miocardiopatía dilatada (MD) y miocardiopatía hipertrófica (MH). Sin embargo, la importancia de la haploinsuficiencia en OBSCN como mecanismo de patogenicidad no está aclarada. El objetivo fue estudiar las variantes en OBSCN que causan haploinsuficiencia y la relación con el fenotipo cardiaco en una cohorte de pacientes analizados con un exoma clínico.
Métodos: De manera retrospectiva se revisaron los análisis genéticos de 714 pacientes índices de un Programa de Cardiopatías Familiares entre marzo de 2019 y marzo de 2022. Los casos fueron analizados mediante secuenciación masiva con un exoma clínico (4.490 genes). Se analizó específicamente la información genética de pacientes portadores de variantes en OBSCN.
Resultados: De todas las variantes no sinónimas detectadas en OBSCN (132 variantes), se identificaron 13 (10%) cuya consecuencia era la haploinsuficiencia en 19 pacientes (tabla), en heterocigosis. De estas, 2 variantes se localizaban en isoforma no cardiaca, 4 eran frameshift, 5 nonsense y 4 de alteración de región canónica de splicing, espaciadas a lo largo del transcrito cardiaco (fig.). El fenotipo predominante de los pacientes era MD. En 8 (42,1%) de estos pacientes se identificó otra variante causal responsable del fenotipo y en 6 (31,6%) casos en los que no se identificó variante causal clara, las variantes en OBSCN identificadas presentaban una frecuencia alélica elevada en población general, por lo que se descartó su causalidad. Al comparar la proporción de variantes no sinónimas identificadas en OBSCN con aquellas identificadas en titina (TTN), se estimó una tasa de variantes en OBSCN muy superior que en TTN lo que sugiere que variantes en OBSCN son mejor toleradas que en TTN, especialmente truncamientos (OBSCN: 0,46 truncamientos/Kb; TTN: 0,20 truncamientos/Kb; p = 0,46).
|
Descripción de variantes que generan truncamiento en el gen OBSCN identificadas en una cohorte de 714 pacientes con miocardiopatía |
|||||||||
|
Paciente |
Gen |
Transcrito |
Isoforma |
cDNA |
Consecuencia |
Proteína |
Fenotipo |
MAF (%) |
Identificación causal |
|
I1 |
OBSCN |
NM_001098623 |
Cardiaca |
c.3768_4043 del |
Alteration of the WT Donor site |
p.(*) |
MS |
0 |
TTN, KCNE2 |
|
I2 |
OBSCN |
NM_001098623 |
Cardiaca |
c.20244+4A> T |
Alteration of the WT Donor site |
p.(*) |
MD |
0 |
SYNE2 VUS+ |
|
I3 |
OBSCN |
NM_001098623 |
Cardiaca |
c.18419+2T> A |
alteration of the WT Donor site |
p.(*) |
MH |
0 |
MYBPC3 |
|
I4 |
OBSCN |
NM_001098623 |
Cardiaca |
c.7126+1G> A |
alteration of The WT Donor site |
p.(*) |
MH+SQTL |
0 |
TTNI3K |
|
I5 |
OBSCN |
NM_001098623 |
Cardiaca |
c.8467A> T |
Nonsense |
p.(Arg2823*) |
MD |
7 |
No causal |
|
I6 |
OBSCN |
NM_001098623 |
Cardiaca |
c.22108G> T |
Nonsense |
p.(Glu7370*) |
MD |
0,1 |
No causal |
|
I7 |
OBSCN |
NM_001098623 |
Cardiaca |
c.11498G> A |
Nonsense |
p.(Trp3833)* |
MAV |
0 |
No causal |
|
I8 |
OBSCN |
NM_001098623 |
Cardiaca |
c.11498G> A |
Nonsense |
p.(Trp3833)* |
MD |
0 |
No causal |
|
I9 |
OBSCN |
NM_001098623 |
Cardiaca |
c.21445G> T |
Nonsense |
p.(Glu7149)* |
MD |
0 |
TNNT2 |
|
I10 |
OBSCN |
NM_001098623 |
Cardiaca |
c.21288 dupT |
Nonsense |
p.(Lys7097) |
MH |
0 |
No causal |
|
I11 |
OBSCN |
NM_001098623 |
Cardiaca |
c.20514_20515 del |
Frameshift |
p.(Ser6839)* |
MD |
0,4 |
MYPN |
|
I12 |
OBSCN |
NM_001098623 |
Cardiaca |
c.20514_20515 del |
Frameshift |
p.(Ser6839)* |
MD |
0,4 |
No causal |
|
I13 |
OBSCN |
NM_001098623 |
Cardiaca |
c.20514_20515 del |
Frameshift |
p.(Ser6839)* |
MD |
0,4 |
MYH7 |
|
I14 |
OBSCN |
NM_001098623 |
Cardiaca |
c.20967 del |
Frameshift |
p.(Ser6990Profs*82) |
MD |
0,2 |
MYH7 |
|
I15 |
OBSCN |
NM_001098623 |
Cardiaca |
c.20967 del |
Frameshift |
p.(Ser6990Profs*82) |
MD |
0,2 |
No causal |
|
I16 |
OBSCN |
NM_001098623 |
Cardiaca |
c.20967 del |
Frameshift |
p.(Ser6990Prof*82) |
MH |
0,2 |
No causal |
|
I17 |
OBSCN |
NM_001098623 |
Cardiaca |
c.20967 del |
Frameshift |
p.(Ser6990Prof*82) |
SQTL |
0,2 |
No causal |
|
I18 |
OBSCN |
NM_001271223 |
No cardiaca |
c.14484_14485 del |
Frameshift |
p.(Val4830Metfs*39) |
MD |
0 |
TTN |
|
I19 |
OBSCN |
NM_001271223 |
No cardiaca |
c.8010 del |
Frameshift |
p.(Thr2672Leufs*31) |
MD |
0 |
RBM20 |
|
MAF: Minor allele frequency (frecuencia del alelo menos común). MS: muerte súbita. MD: miocardiopatía dilatada. MH: miocardiopatía hipertrófica. SQTL: síndrome de QT largo. MAV: miocardiopatía arrimogénica ventricular. |
|||||||||
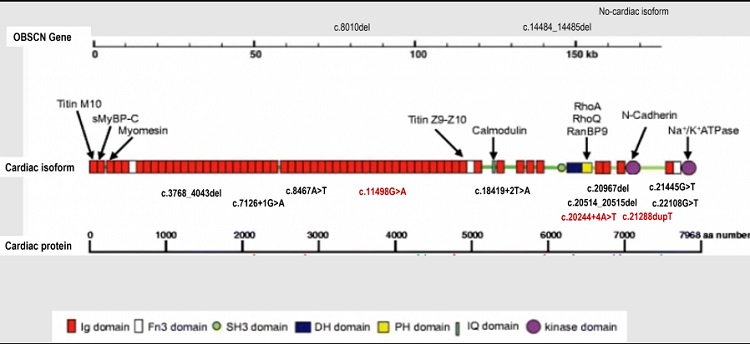
Localización de las variantes en OBSCN que generan truncamientos identificadas en nuestra cohorte (imagen modificada de Fukuzawa et al., 2005).
Conclusiones: Para la mayoría de los probandos portadores de variantes truncantes en OBSCN (74%) se descartó su causalidad. La haploinsuficiencia en OBSCN no constituye un mecanismo principal de patogenicidad en nuestra cohorte de pacientes aunque no se descarta un papel modificador del fenotipo.
Comunicaciones disponibles de "Registros uni y multicéntricos en cardiopatías familiares"
- 4019-1. MODERADOR
- Jesús Piqueras Flores, Ciudad Real
- 4019-2. DETECCIÓN DE ARRITMIAS A LARGO PLAZO MEDIANTE HOLTER INSERTABLE EN PACIENTES CON SÍNDROME DE BRUGADA: RESULTADOS PRELIMINARES DE UN REGISTRO NACIONAL MULTICÉNTRICO
- Eusebio García-Izquierdo1, Julián Palacios Rubio2, Iván Hernández Betancor3, Tomás Ripoll Vera4, Pablo Ramos Ruiz5, Rosa Macías Ruíz6, Ernesto Díaz Infante7, Melodie Segura Domínguez1, Daniel García Rodríguez1, Cristina Aguilera Agudo1, Diego Jiménez Sánchez1, Víctor Castro Urda1, Jorge Toquero Ramos1, Javier Segovia Cubero1 y Ignacio Fernández Lozano1
1Hospital Universitario Puerta de Hierro, Majadahonda, Madrid, 2Hospital Son Espases, Palma de Mallorca, 3Hospital Universitario de Canarias, San Cristóbal de La Laguna, Santa Cruz de Tenerife, 4Hospital Universitario Son Llatzer, Palma de Mallorca, 5Hospital General Universitario Santa Lucía, Cartagena, Murcia, 6Hospital Universitario Virgen de las Nieves, Granada y 7Hospital Universitario Virgen Macarena, Sevilla.
- 4019-3. AFECTACIÓN AÓRTICA Y VASCULAR EN EL SÍNDROME DE LOEYS-DIETZ. RESULTADOS DEL REGISTRO REPAG (RED ESPAÑOLA DE PATOLOGÍA AÓRTICA GENÉTICA)
- Gisela Teixido Tura1, Daniel Martínez2, Francisco Calvo Iglesias3, Rocío García Orta4, Rosario Sánchez5, José María Larrañaga Moreira6, Amparo Hernándiz Martínez7, Elena Díaz8, Elena Montañés9, Fernando Cabrera Bueno10, Anna Sabaté Rotés1, Eduardo Villacorta Argüelles8, J. Francisco Nistal Herrera11, Alberto Forteza Gil2 y Arturo Evangelista Masip1
1Hospital Universitari Vall d'Hebron, Barcelona, 2Hospital Universitario Puerta de Hierro, Majadahonda (Madrid), 3Hospital Álvaro Cunqueiro, Vigo (Pontevedra), 4Hospital Universitario Virgen de las Nieves, Granada, 5Hospital General Universitario de Alicante, 6Complexo Hospitalario Universitario A Coruña, 7Hospital Universitario La Fe, Valencia, 8Hospital Clínico Universitario de Salamanca, 9Hospital Universitario 12 de Octubre, Madrid, 10Hospital Clínico Universitario Virgen de la Victoria, Málaga y 11Hospital Universitario Marqués de Valdecilla, Santander (Cantabria).
- 4019-4. HALLAZGOS FENOTÍPICOS Y GENÉTICOS EN PACIENTES CON SOSPECHA DE ANEURISMA Y DISECCIONES DE LA AORTA TORÁCICA HEREDITARIOS NO SINDRÓMICOS
- Arancha Díaz Expósito, María Victoria Doncel Abad, Daniel Carrasco Gómez, Víctor M. Becerra Muñoz, Patricia Fernández Pérez, José Luis López Benítez, Juan Robledo Carmona, Carlos Porras Martín, José María Melero Tejedor, Manuel Jiménez Navarro, Juan José Gómez Doblas y Fernando Cabrera Bueno
Hospital Clínico Universitario Virgen de la Victoria, Málaga.
- 4019-5. RELEVANCIA DE LAS VARIANTES EN EL GEN OBSCN QUE GENERAN TRUNCAMIENTOS EN PACIENTES CON MIOCARDIOPATÍAS
- María del Carmen García del Rey1, Ana I. Fernández Ávila2, María Ángeles Espinosa1, Silvia Vilches Soria1, Cristina Gómez González1, Irene Méndez Fernández1, Javier Bermejo Thomas1 y Francisco Fernández-Avilés1
1Hospital General Universitario Gregorio Marañón, Madrid y 2CIBERCV. Instituto de Investigación Sanitaria Gregorio Marañón, Madrid.
- 4019-6. ANÁLISIS DE UNA POBLACIÓN CON MIOCARDIOPATÍA ARRITMOGÉNICA SEGÚN SU FORMA DE DEBUT
- Alba María García García1, Juan Ignacio García García2, Gunnar Leithold3, Lidia María Carrillo Mora1, Carmen Muñoz Esparza1, Juan José Sánchez Muñoz1, Cristina Gil Ortuño4, María Sabater Molina4, Noelia Fernández Villa1, Alberto Nieto López1, Manuel Veas Porlán1, David Fernández Vázquez1, José Javier Tercero Fajardo1, Diana Milena Cruz Sepulveda1 y Juan Ramón Gimeno Blanes1
1Hospital Clínico Universitario Virgen de la Arrixaca, Murcia, 2Universidad de Murcia, 3Hospital Virgen del Castillo, Yecla, Murcia y 4Instituto Murciano de Investigación Biosanitaria Virgen de la Arrixaca, Murcia.
- 4019-7. B-MIOSINA Y MIOCARDIOPATÍA HIPERTRÓFICA. NUEVOS HOTSPOTS
- Soledad García-Hernández, Luis de la Higuera Romero, Xusto Fernández, Martín Ortiz Genga, Ivonne J. Cárdenas, María Valverde Gómez, Almudena Amor Salamanca, Laura Cazon Varela, Marlene Pérez Barbeito, Resalía Peteiro Debén, Iria Gómez Díaz, María Sánchez Flores, Anahí Sanluis Verdes y Juan Pablo Ochoa Folmer
Health in Code S.L., A Coruña.
Más comunicaciones de los autores
-
Bermejo Thomas, Javier
- 5006-8 - IMPLANTE DE VÁLVULA AÓRTICA TRANSCATÉTER EN PACIENTES CON ESTENOSIS AÓRTICA GRAVE Y CÁNCER ACTIVO. ANÁLISIS RETROSPECTIVO DE UNA COHORTE UNICÉNTRICA
- 5008-2 - MUERTE SÚBITA CARDIACA NO RECUPERADA. RESULTADOS DE UN PROGRAMA TRANSVERSAL CON AUTOPSIA JUDICIAL, MOLECULAR Y SCREENING FAMILIAR EN UN CENTRO DE REFERENCIA DE CARDIOPATÍAS FAMILIARES
- 5022-8 - CORONARIOGRAFÍA INVASIVA DE ESTRÉS FARMACOLÓGICO CON GUÍA DE PRESIÓN Y ECOGRAFÍA INTRACORONARIA COMO PARTE DE LA ESTRATIFICACIÓN DE RIESGO EN PACIENTES CON ARTERIAS CORONARIAS CON ORIGEN ANÓMALO EN EL SENO CONTRALATERAL. SERIE RETROSPECTIVA EN UN CENTRO TERCIARIO DE REFERENCIA
- 6033-12 - FACTIBILIDAD Y SEGURIDAD DEL ALTA PRECOZ TRAS IMPLANTE MINIMALISTA DE TAVI (FASE-APRETAVI)
- 4021-3 - ESTUDIO DE LA FISIOLOGÍA DEL EJERCICIO MEDIANTE CATETERISMO DE ESFUERZO EN PACIENTES ADULTOS CON CIRCULACIÓN DE FONTAN
- 4019-5 - RELEVANCIA DE LAS VARIANTES EN EL GEN OBSCN QUE GENERAN TRUNCAMIENTOS EN PACIENTES CON MIOCARDIOPATÍAS
-
Espinosa Castro, María Ángeles
- 6030-2 - FENOTIPO Y EVOLUCIÓN CLÍNICA DE AMILOIDOSIS TRANSTIRRETINA HEREDITARIA POR P. GLU109LYS. UNA NUEVA VARIANTE ENDÉMICA EN ESPAÑA
- 4010-2 - PATRONES DE DISTRIBUCIÓN DE REALCE TARDÍO DE GADOLINIO EN MIOCARDIOPATÍA DILATADA NO ISQUÉMICA. CORRELACIÓN GENOTIPO-FENOTIPO
- 5008-2 - MUERTE SÚBITA CARDIACA NO RECUPERADA. RESULTADOS DE UN PROGRAMA TRANSVERSAL CON AUTOPSIA JUDICIAL, MOLECULAR Y SCREENING FAMILIAR EN UN CENTRO DE REFERENCIA DE CARDIOPATÍAS FAMILIARES
- 4019-5 - RELEVANCIA DE LAS VARIANTES EN EL GEN OBSCN QUE GENERAN TRUNCAMIENTOS EN PACIENTES CON MIOCARDIOPATÍAS
-
Fernández Ávila, Ana I.
- 4019-5 - RELEVANCIA DE LAS VARIANTES EN EL GEN OBSCN QUE GENERAN TRUNCAMIENTOS EN PACIENTES CON MIOCARDIOPATÍAS
- 5008-2 - MUERTE SÚBITA CARDIACA NO RECUPERADA. RESULTADOS DE UN PROGRAMA TRANSVERSAL CON AUTOPSIA JUDICIAL, MOLECULAR Y SCREENING FAMILIAR EN UN CENTRO DE REFERENCIA DE CARDIOPATÍAS FAMILIARES
-
Fernández-Avilés, Francisco
- 4002-6 - ESTUDIO DE LA DISFUNCIÓN ENDOTELIAL EN PACIENTES CON ANGINA SIN LESIONES CORONARIAS OBSTRUCTIVAS EN EL REGISTRO NACIONAL ENDOCOR
- 4021-3 - ESTUDIO DE LA FISIOLOGÍA DEL EJERCICIO MEDIANTE CATETERISMO DE ESFUERZO EN PACIENTES ADULTOS CON CIRCULACIÓN DE FONTAN
- 4019-5 - RELEVANCIA DE LAS VARIANTES EN EL GEN OBSCN QUE GENERAN TRUNCAMIENTOS EN PACIENTES CON MIOCARDIOPATÍAS
- 5009-6 - DIFERENCIAS DE GÉNERO EN LA DISFUNCIÓN ENDOTELIAL EN PACIENTES CON ISQUEMIA MIOCÁRDICA SIN LESIONES CORONARIAS OBSTRUCTIVAS EN ESPAÑA
- 6033-12 - FACTIBILIDAD Y SEGURIDAD DEL ALTA PRECOZ TRAS IMPLANTE MINIMALISTA DE TAVI (FASE-APRETAVI)
- 4012-4 - ¿EL IMPLANTE DE ECMO-VA EN HORARIO EXTRALABORAL INFLUYE EN EL PRONÓSTICO DE NUESTROS PACIENTES? EXPERIENCIA DE UN CENTRO TERCIARIO
- 5022-8 - CORONARIOGRAFÍA INVASIVA DE ESTRÉS FARMACOLÓGICO CON GUÍA DE PRESIÓN Y ECOGRAFÍA INTRACORONARIA COMO PARTE DE LA ESTRATIFICACIÓN DE RIESGO EN PACIENTES CON ARTERIAS CORONARIAS CON ORIGEN ANÓMALO EN EL SENO CONTRALATERAL. SERIE RETROSPECTIVA EN UN CENTRO TERCIARIO DE REFERENCIA
- 5024-2 - EXPERIENCIA A 3 MESES DEL USO COMBINADO DE LA PROYECCIÓN DE SOBREPOSICIÓN DE CÚSPIDES Y LA ESTIMULACIÓN AURICULAR EN EL IMPLANTE DE VÁLVULAS AÓRTICAS PERCUTÁNEAS
- 5034-8 - INTELIGENCIA ARTIFICIAL PARA LA DETECCIÓN DE MECANISMOS DE FIBRILACIÓN AURICULAR
- 5024-3 - IMPLANTE DE VÁLVULA AÓRTICA TRANSCATÉTER CON PRÓTESIS AUTOEXPANDIBLE ANULAR EN PACIENTES DE BAJO RIESGO
- 4012-3 - HEMÓLISIS ASOCIADA A SOPORTE CON DISPOSITIVO DE ASISTENCIA VENTRICULAR IZQUIERDA TIPO IMPELLA CP, EXPERIENCIA EN 5 AÑOS EN UN CENTRO TERCIARIO
- 5009-3 - PRONÓSTICO A LARGO PLAZO DEL MANTENIMIENTO DE LA DOBLE TERAPIA ANTIAGREGANTE EN UNA COHORTE DE PACIENTES CONSECUTIVOS CON IAMEST
- 5006-8 - IMPLANTE DE VÁLVULA AÓRTICA TRANSCATÉTER EN PACIENTES CON ESTENOSIS AÓRTICA GRAVE Y CÁNCER ACTIVO. ANÁLISIS RETROSPECTIVO DE UNA COHORTE UNICÉNTRICA
- García del Rey, María del Carmen
-
Gómez González, Cristina
- 4012-4 - ¿EL IMPLANTE DE ECMO-VA EN HORARIO EXTRALABORAL INFLUYE EN EL PRONÓSTICO DE NUESTROS PACIENTES? EXPERIENCIA DE UN CENTRO TERCIARIO
- 5008-2 - MUERTE SÚBITA CARDIACA NO RECUPERADA. RESULTADOS DE UN PROGRAMA TRANSVERSAL CON AUTOPSIA JUDICIAL, MOLECULAR Y SCREENING FAMILIAR EN UN CENTRO DE REFERENCIA DE CARDIOPATÍAS FAMILIARES
- 4019-5 - RELEVANCIA DE LAS VARIANTES EN EL GEN OBSCN QUE GENERAN TRUNCAMIENTOS EN PACIENTES CON MIOCARDIOPATÍAS
- 6030-2 - FENOTIPO Y EVOLUCIÓN CLÍNICA DE AMILOIDOSIS TRANSTIRRETINA HEREDITARIA POR P. GLU109LYS. UNA NUEVA VARIANTE ENDÉMICA EN ESPAÑA
- 6030-7 - IMPACTO DE LA INFECCIÓN POR SARS-COV-2 EN PACIENTES CON AMILOIDOSIS CARDIACA: RESULTADOS DE UN REGISTRO MULTICÉNTRICO INTERNACIONAL
-
Méndez Fernández, Irene
- 4019-5 - RELEVANCIA DE LAS VARIANTES EN EL GEN OBSCN QUE GENERAN TRUNCAMIENTOS EN PACIENTES CON MIOCARDIOPATÍAS
- 5031-5 - MIOCARDIOPATÍA HIPERTRÓFICA POR TRUNCAMIENTOS EN MYOSIN BINDING (MYBPC3): ¿QUÉ PODEMOS ESPERAR?
- 5031-2 - PRONÓSTICO DE LA MIOCARDIOPATÍA HIPERTRÓFICA POR TRUNCAMIENTO EN MYOSIN BINDING (MYBPC3): ¿SIRVEN LAS ESCALAS DE RIESGO CONVENCIONALES?
- 5008-2 - MUERTE SÚBITA CARDIACA NO RECUPERADA. RESULTADOS DE UN PROGRAMA TRANSVERSAL CON AUTOPSIA JUDICIAL, MOLECULAR Y SCREENING FAMILIAR EN UN CENTRO DE REFERENCIA DE CARDIOPATÍAS FAMILIARES
-
Vilches Soria, Silvia
- 5008-2 - MUERTE SÚBITA CARDIACA NO RECUPERADA. RESULTADOS DE UN PROGRAMA TRANSVERSAL CON AUTOPSIA JUDICIAL, MOLECULAR Y SCREENING FAMILIAR EN UN CENTRO DE REFERENCIA DE CARDIOPATÍAS FAMILIARES
- 4019-5 - RELEVANCIA DE LAS VARIANTES EN EL GEN OBSCN QUE GENERAN TRUNCAMIENTOS EN PACIENTES CON MIOCARDIOPATÍAS