ISSN: 0300-8932
Factor de impacto 2024
4,9
SEC 2017 - El Congreso de las Enfermedades Cardiovasculares

Madrid,
26 - 28 de Octubre de 2017
Introducción
Dr. Luis Rodríguez Padial
Presidente del Comité Científico del Congreso
Comité ejecutivo
Comité de evaluadores
Índice de autores
6012. Enfermedades miocardio y pericardio
Fecha
: 26-10-2017 00:00:00
Tipo
: Póster
Sala
: Zona Póster (Planta 3)
6012-204. Síndrome de Carvajal con transmisión autosómica dominante por mutación nonsense en desmoplaquina
Laura Pertejo Manzano, Francisco José Bermúdez Jiménez, Rosa Macías Ruiz, Diego Segura Rodríguez, Emilio Constán de la Revilla, Miguel Álvarez López, Luis Tercedor Sánchez y Juan Jiménez-Jáimez del Servicio de Cardiología del Hospital Virgen de las Nieves, Granada.
Introducción y objetivos: El síndrome de Carvajal es un trastorno cardiocutáneo con patrón de transmisión autosómico recesivo asociado a miocardiopatía arritmogénica de ventrículo derecho (MAVD) con afección mayoritariamente biventricular, queratodermia palmoplantar y pelo “lanudo”, causado por una mutación en el gen desmosómico DSP que codifica la proteína desmoplaquina, esencial en la adhesión celular. Presentamos, por primera vez, una familia con síndrome de Carvajal causado por una única mutación heterocigota R2284X con transmisión autosómica dominante.
Métodos: Estudiamos a una familia a través de un caso índice que debutó con muerte súbita a los 53 años mientras realizaba actividad deportiva, cuya autopsia confirmó MAVD con afección biventricular. En el cribado familiar, sus 2 hijos presentaban criterios compatibles con la enfermedad así como queratodermia palmoplantar, también presente en su progenitor. Se realizó secuenciación de 8 genes responsables de MAVD mediante técnica de Sanger en uno de los descendientes detectándose una única mutación patogénica de tipo nonsense en heterocigosis R2284X/g42476C > T en el exón 24 del gen DSP. El estudio genético en cascada, detectó otros 3 familiares portadores. Realizamos estudio clínico, electrocardiográfico y con técnicas de imagen, así como valoración dermatológica en todos ellos.
Resultados: La mutación DSP R2284X/g42476C > T fue el único hallazgo identificado en el estudio genético del sujeto III-5. El estudio en cascada mostró un patrón de herencia autosómico dominante como muestra el pedigrí (figura). Los no portadores no presentaron clínica, alteraciones ecocardiográficas ni afección cutánea. Entre los portadores, el fenotipo de afección biventricular se observó en el 75% de ellos, mientras que el otro 25% mostró fenotipo de miocardiopatía arritmogénica en la RM, sin cumplir criterios definitivos de la enfermedad (sujeto II-6); todos ellos presentaban alteraciones cutáneas consistentes con queratodermia palmoplantar; En la tabla se analizan más detalladamente las características de los mismos. Tras valorar riesgo de muerte súbita, en los sujetos III-5 y III-6, se optó por implante de DAI en prevención primaria.
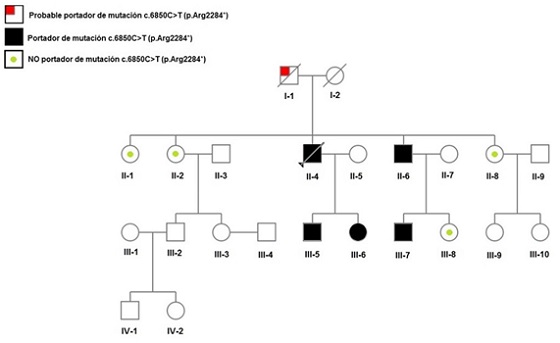
Pedigrí de la familia.
|
Características de los portadores |
|||||||
|
Sujeto |
Clínica |
ECG |
Ecocardiograma |
RMN cardiaca |
Holter |
Ergometría |
Valoración dermatológica |
|
II-6 |
Asintomático |
Normal. |
VI no dilatado con FE normal. VD de diámetros en límite superior con FE normal, sin alteraciones regionales. |
VD ligeramente dilatado en región basal con pared irregular sugestiva de pequeños microaneurismas a este nivel. TSVD ligeramente dilatado. FEVD normal. VI sin alteraciones. Sin realce tardío de gadolinio. |
Sin alteraciones |
No realizada |
Hiperqueratosis ungueal en pies y aislada en algún pulpejo de pies. |
|
Pelo rizado sin alteración aparente del tallo piloso. |
|||||||
|
III-5 |
NYHA I-II. |
RS, bajo voltaje, QRS fragmentado en V2. |
VI ligeramente dilatado con FE ligeramente reducida. VD ligeramente dilatado con FE conservada. |
Infiltración grasa en la porción medio basal de pared anterior de VD. VD ligeramente dilatado con alteraciones de la contractilidad anterior, inferior y apical. Disfunción VD moderada. VI en límite superior de la normalidad, hipocontractilidad global más marcada en porción septal, disfunción ligera de VI. Retención tardía de gadolinio subepicárdica anterior e inferior de VI. |
EV poco frecuente, polimórfica, algunos dobletes muy aislados. Extrasistolia supraventricular muy aislada. |
No realizada |
Hiperqueratosis focal en pulpejos de manos y pies. |
|
Pelo rizado, no se aprecia distrofia de tallo piloso en cuero cabelludo ni cejas |
|||||||
|
III-6 |
NYHA II |
Bajo voltaje generalizado, S fragmentada y ancha en V1-V3. |
Ventrículo izquierdo de diámetros normales-límite superior con FE moderadamente reducida. Ventrículo derecho con FE normal. |
No evidencia de infiltración grasa aparente. VI no dilatado con hipoquinesia inferoseptal y anterior. Disfunción de VI ligera. |
EV de baja densidad, monomorfa. |
9 METs. Muy aislados extrasístoles supraventriculares y ventriculares. |
Eritema difuso palmoplantar con placas más hiperqueratósicas en zonas marginales plantares y pulpejos. |
|
VD no dilatado, hipocinético en región apical, función sistólica en límite inferior de la normalidad. Retención subepicárdica de gadolinio en región lateral basal y media, inferior e inferolateral. |
|||||||
|
III-7 |
Asintomático. |
RS, EV pleomórfica, procedente de VD. Inversión de onda T en cara inferolateral. |
VI no dilatado con FE normal. VD no dilatado con FE normal. No alteraciones regionales significativas apreciables. Diámetro de tracto de salida ligeramente aumentado. |
VI no dilatado con función sistólica en el límite inferior de la normalidad. VD con zonas de microaneurismas en pared libre. Función sistólica ligeramente reducida. |
EV frecuente (> 4.600) con TVNS en pico máximo de esfuerzo de 4 latidos. |
19,3 METs. EV muy frecuente polimórfica durante la prueba. Se registran hasta 14 dupletes. TVNS de 5 latidos. |
Hiperqueratosis focal en área de 6 mm en pulpejos de ambos dedos pulgares. Pelo rizado. No distrofia tallos pilosos en cuero cabelludo y cejas. |
Conclusiones: La mutación en DSP R2284X/g42476C > T se postula como causa de síndrome de Carvajal con patrón de herencia autosómico dominante.
Comunicaciones disponibles de "Enfermedades miocardio y pericardio"
- 6012-190. Validación de una escala de mortalidad a largo plazo en el síndrome de tako-tsubo. RETAKO-score
- Manuel Almendro Delia1, Manuel Lobo González1, Iván Javier Núñez Gil2, Mireia Andrés3, Alessandro Sionis4, Ana Martín5, Teresa Bastante6 y Juan Carlos García Rubira. En Representación de los Investigadores del Registro RETAKO1 del 1Hospital Universitario Virgen Macarena, Sevilla, 2Hospital Clínico San Carlos, Madrid, 3Hospital Universitario Vall d';Hebron, Barcelona, 4Hospital de Sant Pau, Barcelona, 5Hospital Universitario de Salamanca y 6Hospital Universitario de La Princesa, Madrid.
- 6012-191. Predictores de hospitalización por insuficiencia cardiaca en pacientes con miocardiopatía hipertrófica
- Esther González Bartol, Alberto Cecconi, Alberto Vera-Saiz, César Jiménez, Francisco J. de la Cuerda, Jesús Jiménez-Borreguero y Fernando Alfonso del Hospital Universitario de La Princesa, Madrid.
- 6012-192. Influencia pronóstica del sexo en la miocardiopatía hipertrófica por mutación en MYBPC3
- María José Antolinos Pérez, Juan Jiménez Jaimez, María Davo y Luis Tercedor-Sánchez del Hospital Universitario Virgen de las Nieves, Granada.
- 6012-193. Genética en miocardiopatía arritmogénica con expresión fenotípica agresiva, a propósito de un caso
- Carolina Tiraplegui Garjón, Alba Sádaba Cipriain, Gemma Lacuey Lecumberri, Maite Basurte Elorz, Leyre Ucar Rodríguez, Aritza Conty Cardona, Adela María Navarro Echeverria y Lizar Zabala Díaz del Complejo Hospitalario de Navarra, Pamplona (Navarra).
- 6012-194. Diagnóstico clínico en pacientes hospitalizados por pericarditis aguda, ¿cumplimos los criterios diagnósticos?
- Alejandra Ruiz Aranjuelo, Isabel Caballero Jambrina, Javier Jimeno Sánchez, Elena Gambó Ruberte, Ainhoa Pérez Guerrero, Carlos Rubén López Perales, Pablo Auquilla Clavijo e Isabel Calvo Cebollero del Hospital Universitario Miguel Servet, Zaragoza.
- 6012-195. Mutaciones desmosómicas en corazón estructuralmente normal predisponen a miocarditis y muerte súbita
- Óscar Campuzano Larrea1, Georgia Sarquella-Brugada2, Anna Fernández-Falgueras3, Coloma Tirón de Llano3, Alexandra Pérez-Serra3, Josep Castella3, Josep Brugada-Terradellas2 y Ramón Brugada-Terradellas3 del 1Departamento de Ciencias Médicas, Facultad de Medicina, Universidad de Girona, 2Hospital Sant Joan de Déu, Esplugues de Llobregat (Barcelona), y 3Centre de Genètica Cardiovascular, IDIBGI, Universitat de Girona.
- 6012-196. Heterogeneidad en la activación eléctrica en la miocardiopatía arritmogénica a partir del vectocardiograma y el análisis de componentes principales del electrocardiograma
- Jorge Sanz Sánchez1, Santiago Jiménez Serrano2, Begoña Igual Muñoz3, Javier Gimeno Blanes4, Juan Ramón Gimeno Blanes5, José Millet Roig2, Francisco Castells Ramón2 y Esther Zorio Grima1 del 1Hospital Universitario y Politécnico La Fe, IIS La Fe, Valencia, 2Instituto ITACA, Universitat Politècnica de València, 3Hospital General Universitario, Valencia, 4Universidad Miguel Hernández, Murcia, y 5Hospital Clínico Universitario Virgen de la Arrixaca, El Palmar (Murcia).
- 6012-197. Papel del electrocardiograma en el diagnóstico diferencial del dolor torácico no coronario
- Bárbara Serrano Muñoz, Sergio Gamaza Chulián, Rocío Carmona García, Dolores Cañadas Pruaño, Alberto Giráldez Valpuesta y Enrique Otero Chulián del Hospital del S.A.S. de Jerez de la Frontera, Cádiz.
- 6012-198. La alianza entre la patología forense y la cardiología: una ESTRATEGIA útil en la prevención de la muerte súbita cardiaca
- Soledad García Hernández1, Elena Jiménez-Baena1, María Luisa Peña-Peña1, Joaquín Lucena Romero2, Alonso Pedrote Martínez1 y José Eduardo López Haldón1 del 1Hospital Universitario Virgen del Rocío, Sevilla, e 2Instituto de Medicina Legal, Sevilla.
- 6012-199. Análisis de las muertes súbitas por miocardiopatía arritmogénica en nuestro entorno
- Jorge Sanz Sánchez1, M. Paz Suárez-Mier2, Pilar Molina Aguilar3, Beatriz Aguilera-Tapia2, Francisco Pastor Quirante4, Juan Ramón Gimeno Blanes5, Juan Giner Blasco3 y Esther Zorio Grima1 del 1Hospital Universitario y Politécnico La Fe, IIS La Fe, Valencia, 2Instituto Nacional de Toxicología y Ciencias Forenses, Madrid, 3Instituto de Medicina Legal, Valencia, 4Hospital General Universitario Reina Sofía, Murcia, y 5Hospital Clínico Universitario Virgen de la Arrixaca, El Palmar (Murcia).
- 6012-200. La amiloidosis cardiaca no provoca obstrucción en el tracto de salida del ventrículo izquierdo: ¿nueva red flag diagnóstica?
- Sandra Rey-Fariña1, Gonzalo Barge-Caballero2, Juan Martín Loureyro-Rossi3, David Couto-Mallón2, Eduardo Barge-Caballero2, María Jesús Paniagua-Martín2, María G. Crespo-Leiro2 y Roberto Barriales-Villa4 del 1Servicio de Cardiología del Complexo Hospitalario Universitario A Coruña, 2Unidad de Insuficiencia Cardiaca Avanzada y Trasplante Cardiaco del Complexo Hospitalario Universitario A Coruña, 3Servicio de Cardiología del Hospital Provincial del Centenario, Rosario (Argentina), y 4Unidad de Cardiopatías Familiares del Complexo Hospitalario Universitario A Coruña.
- 6012-201. Miocardiopatía hipertrófica apical, una entidad diferente
- Leyre Ucar Rodríguez, Isabel Santos Sánchez, Gemma Lacuey Lecumberri, Carolina Tiraplegui Garjón, Alba Sádaba Cipriain, Maite Basurte Elorz y Virginia Álvarez Asiain del Complejo Hospitalario de Navarra, Pamplona (Navarra).
- 6012-202. Incidencia de la amiloidosis cardiaca como etiología de la hipertrofia miocárdica
- Sonia Ruiz Bustillo, Begoña Benito Villabriga, Helena Tizón Marcos, Nuria Farré López, Aleix Fort Pal, Ruper Oliveró Soldevila, Lluís Molina Ferragut y Julio Martí Almor del Servicio de Cardiología, Hospital del Mar, Barcelona.
- 6012-203. Influencia de la diabetes mellitus en la resolución precoz del edema intramiocárdico en el síndrome de tako-tsubo
- Teresa Alvarado Casas, Paula Antuña Álvarez, Alberto Cecconi, Jorge Salamanca Viloria, Alberto Vera-Saiz, Esther González Bartol, Jesús Jiménez-Borreguero y Fernando Alfonso Manterola del Servicio de Cardiología del Hospital Universitario de La Princesa, Madrid.
- 6012-204. Síndrome de Carvajal con transmisión autosómica dominante por mutación nonsense en desmoplaquina
- Laura Pertejo Manzano, Francisco José Bermúdez Jiménez, Rosa Macías Ruiz, Diego Segura Rodríguez, Emilio Constán de la Revilla, Miguel Álvarez López, Luis Tercedor Sánchez y Juan Jiménez-Jáimez del Servicio de Cardiología del Hospital Virgen de las Nieves, Granada.
- 6012-205. Rentabilidad de la monitorización electrocardiográfica ambulatoria en pacientes con miocardiopatía hipertrófica
- Carlos Anaya Quesada, Rodrigo Di Massa Pezzutti, María Luisa Peña Peña, Julia Rodríguez Ortuño, Elena Jiménez Baena, Antonio J. Ortiz Carrellán, José Eduardo López Haldón y Eva M. Cantero Pérez del Hospital Universitario Virgen del Rocío, Sevilla.
- 6012-206. Factores de riesgo y características de la pericarditis recurrente e incesante en nuestro medio
- Elena Gambó Ruberte, Javier Jimeno Sánchez, Alejandra Ruiz Aranjuelo, Isabel Caballero Jambrina, Ainhoa Pérez Guerrero, Carlos Rubén López Perales, Pablo Esteban Auquilla Clavijo e Isabel Calvo Cebollero del Hospital Universitario Miguel Servet, Zaragoza.
- 6012-207. Patrones electrocardiográficos de una población de pacientes con miocardiopatía hipertrófica
- María Eladia Salar Alcaraz1, Juan Martínez Sánchez2, Pablo Peñafiel Verdú2, Esther Guerrero Pérez2, Juan José Santos Mateo2, Juan Sánchez Serna2, Arcadio García Alberola2 y Juan Ramón Gimeno Blanes2 del 1Hospital General Universitario Los Arcos del Mar Menor, San Javier (Murcia), y 2Hospital Clínico Universitario Virgen de la Arrixaca, El Palmar (Murcia).
Más comunicaciones de los autores
-
Álvarez López, Miguel
- 5008-5 - Predictores de riesgo arrítmico en miocardiopatía arritmogénica entre sexos
- 5006-4 - Tratamiento con flecainida de una mutación en SCN5A asociada a miocardiopatía dilatada y arritmias ventriculares
- 6012-204 - Síndrome de Carvajal con transmisión autosómica dominante por mutación nonsense en desmoplaquina
- 5018-2 - Pacientes con bradiarritmias transitorias relacionadas con fármacos: resultados en el seguimiento
- 6004-87 - Mutaciones missense en filamina c como posible causa de miocardiopatía restrictiva y no compactada
- 6001-14 - Efectividad de la ablación cardiaca en población con edad pediátrica: experiencia de un centro
-
Bermúdez-Jiménez, Francisco José
- 6004-87 - Mutaciones missense en filamina c como posible causa de miocardiopatía restrictiva y no compactada
- 6041-509 - Sacubitrilo/valsartán, experiencia inicial en pacientes de la vida real en una unidad de insuficiencia cardiaca
- 5008-5 - Predictores de riesgo arrítmico en miocardiopatía arritmogénica entre sexos
- 5006-4 - Tratamiento con flecainida de una mutación en SCN5A asociada a miocardiopatía dilatada y arritmias ventriculares
- 6049-591 - Correlación entre perfil lipídico de la dieta y parámetros sanguíneos en pacientes con insuficiencia cardiaca de etiología isquémica y tratamiento hipolipemiante
- 6041-520 - Caracterización clínica de pacientes con insuficiencia cardiaca con fracción de eyección reducida en estadios finales de la vida
- 6012-204 - Síndrome de Carvajal con transmisión autosómica dominante por mutación nonsense en desmoplaquina
- 5018-2 - Pacientes con bradiarritmias transitorias relacionadas con fármacos: resultados en el seguimiento
- 6001-14 - Efectividad de la ablación cardiaca en población con edad pediátrica: experiencia de un centro
- Constán de la Revilla, Emilio
-
Jiménez-Jáimez, Juan
- 5008-5 - Predictores de riesgo arrítmico en miocardiopatía arritmogénica entre sexos
- 6001-14 - Efectividad de la ablación cardiaca en población con edad pediátrica: experiencia de un centro
- 6012-204 - Síndrome de Carvajal con transmisión autosómica dominante por mutación nonsense en desmoplaquina
- 5018-2 - Pacientes con bradiarritmias transitorias relacionadas con fármacos: resultados en el seguimiento
- 6004-87 - Mutaciones missense en filamina c como posible causa de miocardiopatía restrictiva y no compactada
- 5006-4 - Tratamiento con flecainida de una mutación en SCN5A asociada a miocardiopatía dilatada y arritmias ventriculares
- 6012-192 - Influencia pronóstica del sexo en la miocardiopatía hipertrófica por mutación en MYBPC3
- Macías Ruíz, Rosa
-
Pertejo Manzano, Laura
- 6004-87 - Mutaciones missense en filamina c como posible causa de miocardiopatía restrictiva y no compactada
- 6041-525 - Evaluación de la pulsioximetría nocturna como marcador de riesgo en pacientes con insuficiencia cardiaca
- 6012-204 - Síndrome de Carvajal con transmisión autosómica dominante por mutación nonsense en desmoplaquina
- 5006-4 - Tratamiento con flecainida de una mutación en SCN5A asociada a miocardiopatía dilatada y arritmias ventriculares
-
Segura Rodríguez, Diego
- 6012-204 - Síndrome de Carvajal con transmisión autosómica dominante por mutación nonsense en desmoplaquina
- 5008-5 - Predictores de riesgo arrítmico en miocardiopatía arritmogénica entre sexos
- 6013-210 - Análisis de las variables clínicas predictoras del tipo de corrección quirúrgica sobre la válvula tricúspide
- 6004-87 - Mutaciones missense en filamina c como posible causa de miocardiopatía restrictiva y no compactada
-
Tercedor Sánchez, Luis
- 5006-4 - Tratamiento con flecainida de una mutación en SCN5A asociada a miocardiopatía dilatada y arritmias ventriculares
- 6004-87 - Mutaciones missense en filamina c como posible causa de miocardiopatía restrictiva y no compactada
- 6001-14 - Efectividad de la ablación cardiaca en población con edad pediátrica: experiencia de un centro
- 5016-1 - Presentación
- 5018-2 - Pacientes con bradiarritmias transitorias relacionadas con fármacos: resultados en el seguimiento
- 5008-5 - Predictores de riesgo arrítmico en miocardiopatía arritmogénica entre sexos
- 6012-204 - Síndrome de Carvajal con transmisión autosómica dominante por mutación nonsense en desmoplaquina
- 6012-192 - Influencia pronóstica del sexo en la miocardiopatía hipertrófica por mutación en MYBPC3