ISSN: 0300-8932
Factor de impacto 2024
4,9
SEC 2017 - El Congreso de las Enfermedades Cardiovasculares

Madrid,
26 - 28 de Octubre de 2017
Introducción
Dr. Luis Rodríguez Padial
Presidente del Comité Científico del Congreso
Comité ejecutivo
Comité de evaluadores
Índice de autores
6004. Fisiopatología, mecanismos y genética
Fecha
: 26-10-2017 00:00:00
Tipo
: Póster
Sala
: Zona Póster (Planta 3)
6004-87. Mutaciones missense en filamina c como posible causa de miocardiopatía restrictiva y no compactada
Diego Segura Rodríguez, Francisco José Bermúdez-Jiménez, Laura Pertejo Manzano, Rosa Macías Ruíz, José Manuel Oyonarte-Ramírez, Miguel Álvarez-López, Luis Tercedor-Sánchez y Juan Jiménez-Jáimez del Hospital Universitario Virgen de las Nieves, Granada.
Introducción y objetivos: El gen FLNC codifica la filamina C, proteína que se expresa en músculo cardiaco y esquelético. Mutaciones en este gen se han asociado a miopatías esqueléticas y más recientemente se han relacionado con miocardiopatías y muerte súbita. Nuestro objetivo es describir el fenotipo de una serie de casos de miocardiopatías (restrictiva/no compactada) con mutaciones missense en FLNC.
Métodos: Presentamos una serie de 3 pacientes con miocardiopatía primaria (tabla) sin relación de parentesco entre sí, en los que tras realizar estudio genético mediante secuenciación masiva en paralelo se identifica una única mutación, de novo, missense y en heterocigosis en todos los casos, en el gen FLNC.
Resultados: El paciente 1, se trata de un varón de 36 años, diagnosticado de miocardiopatía primaria a los 28 años (solapamiento miocardiopatía no compactada (MNC) y miocardiopatía hipertrófica (MH)), portador de la variante Arg2340Trp. El paciente 2 es un varón de 46 años, diagnosticado de miocardiopatía restrictiva (MR) desde los 30 años, identificándose variante Glu2334Lys. El paciente 3 es un varón de 3 años diagnosticado desde el nacimiento de MNC con patrón restrictivo portador de la variante Pro2301Leu. Se realizó estudio genético de la variante a todos los familiares de primer grado de cada paciente, resultando negativo para la variante missense para FLNC, confirmándola como mutación de novo. La filamina C está formada por una región de unión a actina, 2 regiones bisagra y un dominio con 24 repeticiones de inmunoglobulina-like (figura). Las variantes missense observadas en esta serie, se encuentran en localizadas en la misma inmunoglobulina-like (posición 21) y en aminoácidos muy próximos entre sí (2.300-2.350 aminoácido), lo que puede tener implicación en la patogénesis de la mutación. Aún no existe evidencia suficiente que apoye esta hipótesis, sin embargo, se trata de una variante a tener en cuenta en futuras investigaciones.
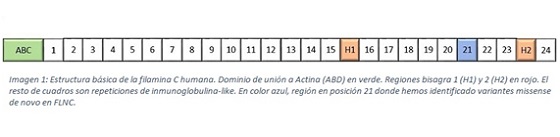
Estructura básica de la filamina C humana.
|
Características clínicas, genéticas y diagnósticas de cada paciente |
|||
|
Paciente |
1 |
2 |
3 |
|
Edad, años |
36 años |
46 años |
3 años |
|
Sexo |
Varón |
Varón |
Varón |
|
Fecha test genético |
20/02/2015 |
02/06/2016 |
11/08/2016 |
|
Gen |
FLNC |
FLNC |
FLNC |
|
Variante |
Arg230Trp |
Glu2334Lys |
Pro2301Leu |
|
Familiares estudiados, n |
3 |
5 |
3 |
|
Familiares afectados, n |
0 |
0 |
0 |
|
Método diagnóstico genético |
Secuenciación masiva |
Secuenciación masiva |
Secuenciación masiva |
|
242 genes |
20 genes |
37 genes |
|
|
Ecocardiografía |
Solapamiento miocardiopatía no compactada/hipertrófica |
Miocardiopatía restrictiva |
Solapamiento miocardiopatía no compactada/restrictiva |
|
Fracción eyección ventrículo izquierdo |
Ligeramente reducida |
Normal |
Normal |
|
Electrocardiograma |
Ritmo sinusal. BCRDHH. |
Fibrilación auricular. BCRDHH. |
Ritmo sinusal. Bloqueo incompleto rama derecha |
|
Descenso ST inferolateral 2 mm |
|||
|
Retención tardía gadolinio(RTG) |
No disponible |
RTG negativo |
No disponible |
|
Holter |
TVNS (4 y 13 latidos) |
Ausencia arritmias ventriculares |
No disponible |
|
CPK (U/L) |
312 |
141 |
No disponible |
|
GGT (U/L) |
60 |
432 |
No disponible |
|
Ergometría |
Buena capacidad funcional. Sin arritmias. |
No realizada |
No realizada |
|
Marcapasos/DAI |
DAI monocameral en prevención primaria |
No |
No |
|
Trasplante cardiaco |
No |
Lista de espera (insuficiencia cardiaca derecha) |
No |
|
BCRDHH: bloqueo completo de rama derecha; RTG: retención tardía gadolinio; TVNS: taquicardia ventricular no sostenida; CPK: creatinafosfoquinasa; GGT: gamma-glutamil-transferasa; DAI: desfibrilador automático implantable. |
|||
Conclusiones: Existen ciertas dudas en la literatura acerca de la patogenicidad de las variantes missense en FLNC. Nuestra serie ilustra que su presencia podría relacionarse con trastornos miocárdicos con expresión fenotípica variada, desde MNC, MR, MH o solapamiento de estas entidades. Dada la escasa evidencia actual, son necesarios más estudios clínicos y funcionales que caractericen mejor estas variantes de la región 21 y su posible patogenicidad.
Comunicaciones disponibles de "Fisiopatología, mecanismos y genética"
- 6004-82. Caracterización de una línea celular de células madre pluripotentes inducidas de un paciente con la transposición de los grandes vasos
- Imelda Ontoria-Oviedo1, Ana María Cervera1, Gábor Földes2, Sandra Tejedor1, Sian E. Harding2, José Anastasio Montero-Argudo1 y Pilar Sepúlveda Sanchís1 de la 1Fundación para la Investigación del Hospital Universitario y Politécnico La Fe, Valencia, y 2National Heart and Lung Institute, Imperial College London, Londres (Reino Unido).
- 6004-83. Prevalencia del bloqueo interauricular avanzado en un población del Norte de Tenerife
- Iván Hernández Betancor1, Juan Lacalzada Almeida2, Javier García-Niebla3, Ignacio Laynez2, Martín Jesús García-González2, Pablo Jorge Pérez2, Patricia Barrio Martínez2 y Antonio Miguel Barragán Acea2 del 1Complejo Hospitalario Universitario de Canarias, San Cristóbal de La Laguna (Tenerife), 2Servicio de Cardiología del Complejo Hospitalario Universitario de Canarias, San Cristóbal de La Laguna (Tenerife), y 3Centro de Salud Valle del Golfo, Frontera (Tenerife).
- 6004-84. Taquicardia ventricular polimórfica catecolaminérgica y riesgo de muerte súbita en el contexto de una nueva deleción de 3 exones en CASQ2
- María Brion 1, Alejandro Blanco-Verea1, Eva Ramos-Luis1, María Álvarez-Barredo2, Moises Rodríguez-Mañero2, Ángel Fernández-López2, Ángel Carracedo3 y José Ramón González-Juanatey2 de 1Xenética Cardiovascular, Instituto de Investigación Sanitaria Santiago de Compostela (IDIS), A Coruña, 2Servicio de Cardiología, Complexo Hospitalario Universitario de Santiago, Santiago de Compostela (A Coruña), y 3Fundación Pública Galega de Medicina Xenómica, Santiago de Compostela (A Coruña).
- 6004-85. Indicación de estudio genético en menores de edad
- Óscar Campuzano Larrea1, Anna Fernández-Falgueras2, Georgia Sarquella-Brugada3, Sergi César3, Elena Arbelo3, Fernando Wangüemert2, Josep Brugada-Terradellas3 y Ramón Brugada-Terradellas2 del 1Departamento Ciencias Médicas, Facultad Medicina, Universidad de Girona, 2Centre de Genètica Cardiovascular, IDIBGI, Universitat de Girona y 3Hospital Sant Joan de Déu, Esplugues de Llobregat (Barcelona).
- 6004-86. Relación entre el intervalo pico-fin de la onda T y choques apropiados en pacientes con síndrome de Brugada y cardiodesfibriladores implantables
- Alejo Tronconi, Néstor Galizio, Gabriela Lizarraga, Fernanda Figueroa, Federico Robles, Alejandro Palazzo, Guillermo Carnero y José Luis González del Hospital Universitario Fundación Favaloro, Buenos Aires (Argentina).
- 6004-87. Mutaciones missense en filamina c como posible causa de miocardiopatía restrictiva y no compactada
- Diego Segura Rodríguez, Francisco José Bermúdez-Jiménez, Laura Pertejo Manzano, Rosa Macías Ruíz, José Manuel Oyonarte-Ramírez, Miguel Álvarez-López, Luis Tercedor-Sánchez y Juan Jiménez-Jáimez del Hospital Universitario Virgen de las Nieves, Granada.
- 6004-88. Muerte súbita en cirugía univentricular, ¿cómo de frecuente? ¿cómo de predecible?
- Inmaculada Sánchez Pérez, Natalia Rivero Jiménez, Javier Moreno Planas, Eduardo Franco Díez, Roberto Matía Francés, Antonio Hernández Madrid, Vanesa Cristina Lozano Granero y María Jesús del Cerro Marín del Hospital Universitario Ramón y Cajal, Madrid.
- 6004-89. Metformina previene la fibrosis cardiaca tras infarto de miocardio, a través de la atenuación de la ruta de señalización NADPH oxidasa/PKCa/Gal-3
- Juan José Santos Mateo1, Domingo Pascual-Figal1, María Teresa Pérez-Martínez2, María del Carmen Sánchez-Pérez2, Yassine Sassi3, Roger Hajjar3, Antonio Lax Pérez2 y María del Carmen Asensio López2 del 1Hospital Clínico Universitario Virgen de la Arrixaca, El Palmar (Murcia), 2Instituto Murciano de Investigación Biosanitaria Virgen de la Arrixaca (IMIB-Arrixaca), Murcia, y 3Icahn School of Medicine at Mount Sinai, New York (Estados Unidos).
- 6004-90. Mapeo simultáneo endoepicárdico durante estiramiento global en corazón aislado revela propiedades de restitución bifásicas responsables del incremento de complejidad durante fibrilación ventricular
- Conrado J. Calvo Saiz1, Álvaro Tormos1, Eduardo Roses1, José Millet1, Luis Such-Miquel2, Manolo Zarzoso2, Javier Chorro2 y Antonio Guill1 del 1Instituto ITACA, Universitat Politècnica de València y 2Universidad de Valencia.
- 6004-91. Varón con infarto inferior: alerta TV
- Luis Borrego Bernabé1, Iván Javier Núñez Gil1, Eduardo Pozo Osinalde1, Julián Palacios Rubio1, David Filgueiras Rama1, Nicasio Pérez Castellano1, Juan Carlos García Rubira2 y Julián Pérez-Villacastín3 de 1Cardiología, Hospital Clínico San Carlos, Madrid, 2Cardiología, Hospital Universitario Virgen Macarena, Sevilla, y 3Hospital Clínico San Carlos, Madrid, CIBER de Enfermedades Cardiovasculares.
Más comunicaciones de los autores
-
Álvarez López, Miguel
- 5008-5 - Predictores de riesgo arrítmico en miocardiopatía arritmogénica entre sexos
- 5006-4 - Tratamiento con flecainida de una mutación en SCN5A asociada a miocardiopatía dilatada y arritmias ventriculares
- 6012-204 - Síndrome de Carvajal con transmisión autosómica dominante por mutación nonsense en desmoplaquina
- 5018-2 - Pacientes con bradiarritmias transitorias relacionadas con fármacos: resultados en el seguimiento
- 6001-14 - Efectividad de la ablación cardiaca en población con edad pediátrica: experiencia de un centro
- 6004-87 - Mutaciones missense en filamina c como posible causa de miocardiopatía restrictiva y no compactada
-
Bermúdez-Jiménez, Francisco José
- 6004-87 - Mutaciones missense en filamina c como posible causa de miocardiopatía restrictiva y no compactada
- 6041-509 - Sacubitrilo/valsartán, experiencia inicial en pacientes de la vida real en una unidad de insuficiencia cardiaca
- 5008-5 - Predictores de riesgo arrítmico en miocardiopatía arritmogénica entre sexos
- 5006-4 - Tratamiento con flecainida de una mutación en SCN5A asociada a miocardiopatía dilatada y arritmias ventriculares
- 6049-591 - Correlación entre perfil lipídico de la dieta y parámetros sanguíneos en pacientes con insuficiencia cardiaca de etiología isquémica y tratamiento hipolipemiante
- 6041-520 - Caracterización clínica de pacientes con insuficiencia cardiaca con fracción de eyección reducida en estadios finales de la vida
- 6012-204 - Síndrome de Carvajal con transmisión autosómica dominante por mutación nonsense en desmoplaquina
- 5018-2 - Pacientes con bradiarritmias transitorias relacionadas con fármacos: resultados en el seguimiento
- 6001-14 - Efectividad de la ablación cardiaca en población con edad pediátrica: experiencia de un centro
-
Jiménez-Jáimez, Juan
- 6012-192 - Influencia pronóstica del sexo en la miocardiopatía hipertrófica por mutación en MYBPC3
- 6004-87 - Mutaciones missense en filamina c como posible causa de miocardiopatía restrictiva y no compactada
- 6001-14 - Efectividad de la ablación cardiaca en población con edad pediátrica: experiencia de un centro
- 5018-2 - Pacientes con bradiarritmias transitorias relacionadas con fármacos: resultados en el seguimiento
- 5008-5 - Predictores de riesgo arrítmico en miocardiopatía arritmogénica entre sexos
- 5006-4 - Tratamiento con flecainida de una mutación en SCN5A asociada a miocardiopatía dilatada y arritmias ventriculares
- 6012-204 - Síndrome de Carvajal con transmisión autosómica dominante por mutación nonsense en desmoplaquina
- Macías Ruíz, Rosa
- Oyonarte-Ramírez, José Manuel
-
Pertejo Manzano, Laura
- 6041-525 - Evaluación de la pulsioximetría nocturna como marcador de riesgo en pacientes con insuficiencia cardiaca
- 5006-4 - Tratamiento con flecainida de una mutación en SCN5A asociada a miocardiopatía dilatada y arritmias ventriculares
- 6012-204 - Síndrome de Carvajal con transmisión autosómica dominante por mutación nonsense en desmoplaquina
- 6004-87 - Mutaciones missense en filamina c como posible causa de miocardiopatía restrictiva y no compactada
-
Segura Rodríguez, Diego
- 6004-87 - Mutaciones missense en filamina c como posible causa de miocardiopatía restrictiva y no compactada
- 5008-5 - Predictores de riesgo arrítmico en miocardiopatía arritmogénica entre sexos
- 6013-210 - Análisis de las variables clínicas predictoras del tipo de corrección quirúrgica sobre la válvula tricúspide
- 6012-204 - Síndrome de Carvajal con transmisión autosómica dominante por mutación nonsense en desmoplaquina
-
Tercedor Sánchez, Luis
- 6004-87 - Mutaciones missense en filamina c como posible causa de miocardiopatía restrictiva y no compactada
- 6012-192 - Influencia pronóstica del sexo en la miocardiopatía hipertrófica por mutación en MYBPC3
- 5006-4 - Tratamiento con flecainida de una mutación en SCN5A asociada a miocardiopatía dilatada y arritmias ventriculares
- 6001-14 - Efectividad de la ablación cardiaca en población con edad pediátrica: experiencia de un centro
- 5008-5 - Predictores de riesgo arrítmico en miocardiopatía arritmogénica entre sexos
- 5016-1 - Presentación
- 5018-2 - Pacientes con bradiarritmias transitorias relacionadas con fármacos: resultados en el seguimiento
- 6012-204 - Síndrome de Carvajal con transmisión autosómica dominante por mutación nonsense en desmoplaquina