ISSN: 0300-8932
Factor de impacto 2024
4,9
SEC 2017 - El Congreso de las Enfermedades Cardiovasculares

Madrid,
26 - 28 de Octubre de 2017
Introducción
Dr. Luis Rodríguez Padial
Presidente del Comité Científico del Congreso
Comité ejecutivo
Comité de evaluadores
Índice de autores
5008. Novedades en miocardiopatías
Fecha
: 26-10-2017 15:15:00
Tipo
: Comunicaciones mini orales
Sala
: Sala La Paz (Planta 2. Dcha.)
5008-8. Diferentes manifestaciones de una misma enfermedad: análisis de la variación fenotípica en varias familias con la misma mutación en troponina I
Fernando Wanguemert Pérez1, Federico Segura Villalobos2, María del Val Groba Marco3, Antonio García Quintana3, Juan Carlos Rodríguez Pérez2, Eduardo Caballero Dorta3 y José María Medina Gil2 del 1Cardiavant Centro Médico Cardiológico, Las Palmas de Gran Canaria, 2Complejo Hospitalario Universitario Insular Materno Infantil de Gran Canaria, Las Palmas de Gran Canaria, y 3Hospital Universitario de Gran Canaria Doctor Negrín, Las Palmas de Gran Canaria.
Introducción y objetivos: Aproximadamente el 2-7% de los casos de miocardiopatía familiar son causados por una mutación en el gen que codifica la troponina I cardiaca (TNNI3). La mayoría de estas mutaciones se encuentran en pacientes con miocardiopatía hipertrófica (MCH) (85%), restrictiva (MCR) (8%) y rara vez en miocardiopatías dilatadas (MCD). El objetivo del presente estudio fue describir el abordaje diagnóstico y las manifestaciones fenotípicas de 3 familias portadoras de la misma mutación p.Arg186Gln en TNNI3.
Métodos: Se estudiaron 3 casos índices de manera independiente, sin relación familiar establecida, en 3 centros diferentes (figura). Todos los probandos tenían realizado un electrocardiograma, ecocardiografía transtorácica, resonancia magnética (RM) y estudio genético. El estudio genético reveló la mutación en heterocigosis p.Arg186Gln en TNNI3. Se comparó el fenotipo y las manifestaciones clínicas de los probandos así como en los familiares.
Resultados: En la tabla se presentan los datos clínicos, edad, datos ecográficos y presencia de realce tardío en la resonancia de los probandos de las 3 familias. La misma mutación, tiene un comportamiento clínico y fenotípico distinto: En la familia I predomina la MCH y la muerte súbita, en la familia II, sin embargo hay al menos 2 casos de MCR con insuficiencia cardiaca terminal, y en la familia III predomina la MCH. Se observa una alta penetrancia de la enfermedad, y una manifestación clínica heterogénea. A pesar de la probable relación entre las 3 familias, no hemos podido vincularlas en el árbol familiar en la actualidad.
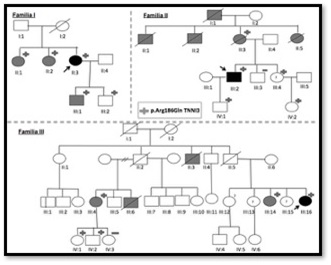
Representación del árbol genealógico de las familias I, II y III.
|
Datos disponibles de los probando de las 3 familias |
||||
|
Familia |
Edad diagnóstico |
Presentación clínica |
Ecocardiograma: grosor máximo pared |
Resonancia: realce tardío |
|
I-II:3 |
55 |
Fibrilación auricular |
M. hipertrófica 19 mm |
Sí |
|
I-II:1 |
51 |
Muerte súbita recuperada |
M. hipertrófica 19 mm |
Sí |
|
I-III:1 |
20 |
Muerte súbita recuperada |
M. hipertrófica 22 mm |
Sí |
|
I-II:2 |
49 |
Asintomática |
M. hipertrófica 22 mm |
No realizada |
|
II-III:2 |
40 |
Asintomático |
14 mm |
No realce |
|
II-III:4 |
42 |
Asintomática |
Normal |
No realizada |
|
II-II:3 |
58 |
I. cardiaca |
M. restrictiva |
No disponible |
|
II-II:5 |
48 |
Muerte súbita |
M. hipertrófica-restrictiva |
No disponible |
|
II-II:1 |
39 |
I. cardiaca terminal |
M. restrictiva |
No disponible |
|
II-II:2 |
37 |
Muerte súbita |
No disponible |
No disponible |
|
II-IV:1 |
8 |
Asintomático |
Normal |
No realizada |
|
III-III:16 |
40 |
Asintomática |
M. hipertrófica 15 mm |
Sí |
|
III-III:14 |
47 |
Asintomática |
M. hipertrófica 20 mm |
Sí |
|
III-III:6 |
36 |
Muerte súbita |
M. hipertrófica? |
No disponible |
|
III-III:4 |
64 |
I. cardiaca |
M. hipertrófica 22 mm |
Sí |
|
III-II:3 |
50 |
Muerte súbita |
? |
No disponible |
|
III-IV:1 |
31 |
Asintomática |
Normal |
No realizada |
|
III-IV:2 |
42 |
Asintomático |
Normal |
No realizada |
Conclusiones: Presentamos varias formas de enfermedad de la misma mutación p.Arg186Gln en TNNI3, en 3 familias aparentemente independientes, pero con un probable ancestro común. Destacamos la importancia del cribado genético para diagnosticar a los miembros de la familia asintomáticos o con un fenotipo sutil, que también presentan un alto riesgo de muerte súbita y por lo tanto también necesitan un seguimiento más específico. Finalmente señalar la necesidad de una fuerte colaboración entre los distintos centros para completar los estudios familiares de enfermedades genéticas como esta, y así analizar de forma conjunta el comportamiento de la enfermedad, y las alternativas terapéuticas.
Comunicaciones disponibles de "Novedades en miocardiopatías"
- 5008-1. Presentación
- Francisco Torres Calvo, Málaga, y Ana García Martín, Madrid.
- 5008-2. Correlación genotipo-fenotipo en no compactación miocárdica del ventrículo izquierdo
- Guillem Casas Masnou, Gerard Oristrell Santamaría, Mar Borregan, Laura Gutiérrez García-Moreno, Javier Limeres Freire, Arturo Evangelista Masip, David García Dorado y José Fernando Rodríguez Palomares del Hospital Universitario Vall d'Hebron, Barcelona.
- 5008-3. Registro español de cardiolaminopatías (REDLAMINA)
- Joel Salazar-Mendiguchía1, Pablo García-Pavía2, María Luisa Peña-Peña3, Tomás Ripoll-Vera4, Esther Zorio-Grima5, Vicente Climent-Payá6, Raquel Yotti-Álvarez7 y Roberto Barriales-Villa8 del 1Departamento Clínico de Healthincode, A Coruña, 2Unidad de Cardiopatías Familiares del Hospital Puerta de Hierro, Madrid, 3Unidad de Cardiopatías Familiares del Hospital Universitario Virgen del Rocío, Sevilla, 4Unidad de Cardiopatías Familiares del Hospital Universitario Son Llàtzer, Palma de Mallorca, 5Servicio de Cardiología del Hospital Universitario La Fe, Valencia, 6Servicio de Cardiología, Hospital General Universitario de Alicante, Alicante, 7Servicio de Cardiología, Hospital General Universitario Gregorio Marañón, Madrid, y 8Unidad de Cardiopatías Familiares del Complexo Hospitalario Universitario A Coruña, A Coruña.
- 5008-4. Predictores de muerte súbita en pacientes con laminopatía congénita. La importancia de la evaluación cardiológica sistematizada precoz
- Georgia Sarquella-Brugada1, Sergi César1, Carles Bautista1, Isaac Moll1, Andrés Nascimento2, Carlos Ortez2, Oscar Campuzano3 y Josep Brugada4 de la 1Unidad de Arritmias y Muerte Súbita, Hospital Sant Joan de Déu, Esplugues de Llobregat (Barcelona), 2Unidad de Neuromuscular, Hospital Sant Joan de Déu, Esplugues de Llobregat (Barcelona), 3Centre de Genètica Cardiovascular, IDIBGI, Universitat de Girona, y 4Unidad de Arritmias, Hospital Sant Joan de Déu, Esplugues de Llobregat (Barcelona).
- 5008-5. Predictores de riesgo arrítmico en miocardiopatía arritmogénica entre sexos
- Francisco José Bermúdez Jiménez, Erika López Moreno, Diego Segura-Rodríguez, Silvia López-Fernández, Miguel Álvarez-López, Luis Tercedor-Sánchez, Juan Jiménez-Jáimez y Concepción Correa-Vílchez del Complejo Hospitalario Universitario de Granada.
- 5008-6. Miocardiopatía hipertrófica obstructiva tratada con marcapasos biventricular y ablación de nodo AV-Seguimiento a largo plazo
- Markus Linhart, Ada Doltra, Diego Penela, Mikel Martínez, Felip Burgos, Juan Acosta, Lluís Mont y Antonio Berruezo del Hospital Clínic, Departamento de Cardiología, Barcelona.
- 5008-7. Utilidad de la imagen combinada y la biopsia extracardiaca en el diagnóstico no invasivo de la amiloidosis cardiaca
- Rodolfo Antonio Montiel Quintero, María del Val Groba Marco, Aridane Cárdenes León, Irene Menduiña Gallego, Marta Blanco Nuez, Marta López Pérez, Antonio García Quintana y Eduardo José Caballero Dorta del Hospital Universitario de Gran Canaria Doctor Negrín, Las Palmas de Gran Canaria.
- 5008-8. Diferentes manifestaciones de una misma enfermedad: análisis de la variación fenotípica en varias familias con la misma mutación en troponina I
- Fernando Wanguemert Pérez1, Federico Segura Villalobos2, María del Val Groba Marco3, Antonio García Quintana3, Juan Carlos Rodríguez Pérez2, Eduardo Caballero Dorta3 y José María Medina Gil2 del 1Cardiavant Centro Médico Cardiológico, Las Palmas de Gran Canaria, 2Complejo Hospitalario Universitario Insular Materno Infantil de Gran Canaria, Las Palmas de Gran Canaria, y 3Hospital Universitario de Gran Canaria Doctor Negrín, Las Palmas de Gran Canaria.
Más comunicaciones de los autores
-
Caballero Dorta, Eduardo
- 6008-131 - Supervivencia a largo plazo en pacientes sometidos a desfibrilador resincronizador frente a marcapasos resincronizador. Seguimiento a largo plazo
- 6041-514 - ¿El marcapasos resincronizador puede tener un papel importante según el tipo de miocardiopatía? Estudio en una cohorte prospectiva a largo plazo
- 5008-7 - Utilidad de la imagen combinada y la biopsia extracardiaca en el diagnóstico no invasivo de la amiloidosis cardiaca
- 6002-30 - Terapia de resincronización cardiaca: upgrade frente a novo. Seguimiento a largo plazo
- 5008-8 - Diferentes manifestaciones de una misma enfermedad: análisis de la variación fenotípica en varias familias con la misma mutación en troponina I
- 7001-18 - Cardiopatía isquémica y upgrade de dispositivo de terapia de resincronización cardiaca
- 6002-33 - ¿Se debe optar siempre por implantar un desfibrilador resincronizador respecto al marcapasos resincronizador? Estudio comparativo a largo plazo
- 6041-508 - ¿Es necesario el upgrade en pacientes sometidos a terapia de resincronización cardiaca?
- 6008-132 - ¿Es efectivo el upgrade en los pacientes con insuficiencia cardiaca? Seguimiento a largo plazo
-
García Quintana, Antonio
- 5008-8 - Diferentes manifestaciones de una misma enfermedad: análisis de la variación fenotípica en varias familias con la misma mutación en troponina I
- 5008-7 - Utilidad de la imagen combinada y la biopsia extracardiaca en el diagnóstico no invasivo de la amiloidosis cardiaca
- 6002-33 - ¿Se debe optar siempre por implantar un desfibrilador resincronizador respecto al marcapasos resincronizador? Estudio comparativo a largo plazo
- Groba Marco, María del Val
-
Medina Gil, José María
- 5008-8 - Diferentes manifestaciones de una misma enfermedad: análisis de la variación fenotípica en varias familias con la misma mutación en troponina I
- 6015-235 - ¿Es el ratio plaquetas/linfocitos útil como variable predictora de la gravedad del síndrome coronario agudo?
- 6015-234 - Evaluación del ratio neutrófilos/linfocitos como variable inflamatoria predictora de la gravedad del síndrome coronario agudo
- Rodríguez Pérez, Juan Carlos
- Segura Villalobos, Federico
- Wanguemert Pérez, Fernando