ISSN: 0300-8932
Factor de impacto 2024
4,9
SEC 2012 - El Congreso de las Enfermedades Cardiovasculares

Sevilla,
18 - 20 de Octubre de 2012
4008. Cardiología pediátrica-Cardiopatías congénitas II
Fecha
: 18-10-2012 00:00:00
Tipo
: Comunicaciones mini orales
Sala
: Sala C1 (Planta 1)
4008-3. Hipertensión pulmonar asociada a cardiopatías congénitas y shunt sistémico pulmonar: ¿es siempre mejor cerrar? Datos del Registro Español de Hipertensión Pulmonar (REHAP)
Carmen Jiménez López-Guarch, Laura Dos Subirà, José María Oliver, Javier Segovia Cubero, María Lázaro Salvador, Maite Subirana Doménech, María Quero y Pilar Escribano Subias del Registro Español de Hipertensión Pulmonar (REHAP), Madrid, Registro Español de Hipertensión Pulmonar (REHAP), Barcelona y Registro Español de Hipertensión Pulmonar (REHAP), Toledo.
Introducción: La hipertensión pulmonar asociada a cardiopatías congénitas (HAP-CC) se desarrolla en un subgrupo pacientes con un shunt sistémico pulmonar tras un tiempo de latencia, que depende del defecto (tamaño, localización). Existen escasos datos clínicos y pronósticos sobre el subgrupo de pacientes en los que se diagnostica HAP tras el cierre quirúrgico o percutáneo del defecto. El registro español de hipertensión pulmonar (REHAP) se inicio en 2007, e incluye a todas las formas de HAP, entre ellas pacientes con HAP-CC. El objetivo del presente estudio es analizar las características clínicas y pronósticas de los pacientes con HAP-CC tras la corrección de defecto, sin shunt residual, y compararlo con el grupo más numeroso y conocido, los pacientes con síndrome de Eisenmenger.
Métodos: Inclusión voluntaria de pacientes (pac.) > 14 años con HAP-CC diagnosticada mediante cateterismo derecho. Los 4 grandes centros de referencia de HP y CC incluyeron el 75% de los pac. que se estratificaron en los 2 grupos de la nueva clasificación: 1) Eisenmenger; 2) HAP con defecto corregido sin shunt residual. Se analizaron las diferencias demográficas, clínicas, hemodinámicas y de supervivencia.
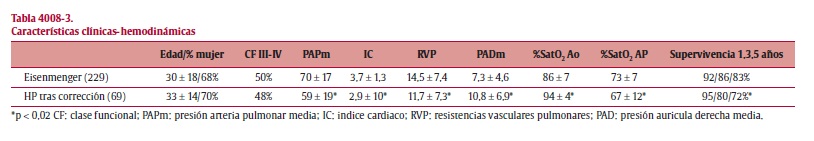
Resultados: Se incluyeron 298 pacientes (18% de la población total del REHAP). Las diferencias entre grupos se expresan en la tabla.
Conclusiones: 1) El REHAP supone el 1er registro de HAP-CC en nuestro país y uno de los más numerosos a nivel Europeo. 2) Los pacientes con defecto corregido tienen menor PAPm, y sin embargo asocian datos de peor función de VD: menor índice cardiaco, y saturación de O2 en AP y mayor PADm. 3) La supervivencia es significativamente peor en este grupo con respecto al S. Eisenmenger. 4) Se debe establecer unos criterios muy estrictos para decidir el cierre de defectos en los pacientes con HP y shunt sistémico-pulmonares, ya que la decisión inadecuada de cierre puede conllevar un pronóstico marcadamente adverso.
Comunicaciones disponibles de "Cardiología pediátrica-Cardiopatías congénitas II"
- 4008-1. Presentación
- José M. Oliver Ruiz, Madrid y José Luis Zunzunegui, Madrid.
- 4008-2. Capacidad funcional aeróbica en adultos con anomalía de Ebstein no intervenida
- Irene Méndez Santos, Pablo Bastos Amador, María Rocío Gómez Domínguez, Blanca Muñoz Calero, Luis González Torres, María Luisa Cabeza Letrán, María José Rodríguez Puras y Pastora Gallego García de Vinuesa del Área del Corazón (Cardiopatías Congénitas del adulto), Hospital Universitario Virgen Macarena y Hospital Universitario Virgen del Rocío, Sevilla.
- 4008-3. Hipertensión pulmonar asociada a cardiopatías congénitas y shunt sistémico pulmonar: ¿es siempre mejor cerrar? Datos del Registro Español de Hipertensión Pulmonar (REHAP)
- Carmen Jiménez López-Guarch, Laura Dos Subirà, José María Oliver, Javier Segovia Cubero, María Lázaro Salvador, Maite Subirana Doménech, María Quero y Pilar Escribano Subias del Registro Español de Hipertensión Pulmonar (REHAP), Madrid, Registro Español de Hipertensión Pulmonar (REHAP), Barcelona y Registro Español de Hipertensión Pulmonar (REHAP), Toledo.
- 4008-4. Uso profiláctico de esteroides en niños con cardiopatías congénitas operados con apoyo de circulación extracorpórea
- Hungría Fernández González, Eduardo Romero Veccione e Isabel Iturria Caamaño del Hospital Cardiológico Infantil Latinoamericano Dr. Gilberto Rodríguez Ochoa, Caracas y Universidad Central de Venezuela, Caracas.
- 4008-5. Estudio de la morbimortalidad en pacientes con d-transposición de grandes arterias (DTGA) sometidos a cirugía de recambio arterial
- María José Rodríguez Puras, María Luisa Cabeza Letrán, Manuela Romero-Vazquianez, José Santos de Soto, Antonio Álvarez Madrid, Reza Housseinpour, Irene Méndez Santos y Pastora Gallego García de Vinuesa del Área del Corazón (Cardiopatías Congénitas del adulto) del Hospital Universitario Virgen Macarena y Hospital Universitario Virgen del Rocío, Sevilla.
- 4008-6. Influencia del fenotipo de válvula bicúspide en la coartación de aorta
- María Rocío Gómez Domínguez, Luis González Torres, Blanca Muñoz Calero, Irene Méndez Santos, Carlos Caparrós Escudero, Sergio Rodríguez de Leiras, Marinela Chaparro Muñoz y Pastora Gallego García de Vinuesa del Área del Corazón, Hospital Universitario Virgen Macarena y Hospital Universitario Virgen del Rocío, Sevilla.
- 4008-7. Impacto del embarazo en mujeres con D-transposición de grandes arterias sometidas a switch atrial
- Valle Pedrosa del Moral, Antònia Pijuan Doménech, Laura Dos Subirà, María Goya Canino, Francesc Baró Mariné, María Carmen Merced Vázquez, Maite Subirana Doménech y Jaume Casaldàliga-Ferrer de la Unidad de Cardiopatías Congénitas del Adolescente y del Adulto (UCCAA), Servicio de Ginecología y Obstetricia del Hospital General Universitario Vall d'Hebron, Barcelona y Hospital de la Santa Creu i Sant Pau, Barcelona.
- 4008-8. Cierre percutáneo de dehiscencias en las neoaurículas en pacientes con cirugía de Switch auricular
- Andreu Porta-Sánchez, Gerard Martí-Aguasca, Josep Girona Comas, M. Antònia Pijuan-Domènech, Laura Dos-Subirà, Maite Subirana Doménech y Jaume Casaldàliga-Ferrer del Servei de Cardiologia, Servei de Cardiologia Pediàtrica y Unitat de Cardiopaties Congènites de l'Adult del Hospital Universitari Vall d'Hebron, Barcelona.
Más comunicaciones de los autores
-
Dos Subirà, Laura
- 4008-7 - Impacto del embarazo en mujeres con D-transposición de grandes arterias sometidas a switch atrial
- 6001-394 - Hipertensión pulmonar en adultos con cardiopatía congénita
- 4008-3 - Hipertensión pulmonar asociada a cardiopatías congénitas y shunt sistémico pulmonar: ¿es siempre mejor cerrar? Datos del Registro Español de Hipertensión Pulmonar (REHAP)
- 6001-417 - Accidentes cerebrovasculares de etiología embólica en adultos con tetralogía de fallot. estudio multicéntrico retrospectivo
-
Escribano Subias, Pilar
- 4048-12 - Prevención en hipertensión pulmonar. ¿cómo estamos tratando a nuestros pacientes?
- 6000-45 - ¿Cuál es el perfil del paciente con hipertensión pulmonar precapilar en España? Estudio multicéntrico
- 6000-200 - El índice de trabajo sistólico del ventrículo derecho predice mortalidad después de tromboendarterectomía en pacientes con hipertensión pulmonar tromboembólica crónica
- 4008-3 - Hipertensión pulmonar asociada a cardiopatías congénitas y shunt sistémico pulmonar: ¿es siempre mejor cerrar? Datos del Registro Español de Hipertensión Pulmonar (REHAP)
- 4045-4 - La infección por citomegalovirus tiene una influencia decisiva en el desarrollo de enfermedad vascular del injerto
- 4027-6 - Arteriografía pulmonar en pacientes con hipertensión pulmonar tromboembólica crónica como parte de la evaluación previa a cirugía de tromboendarterectomía pulmonar. Tolerancia y seguridad
- 4027-8 - El índice de trabajo sistólico del ventrículo derecho predice mortalidad en pacientes con hipertensión pulmonar tromboembólica crónica desestimados para cirugía
- 6000-202 - El índice de trabajo sistólico del ventrículo derecho se correlaciona con los parámetros ecocardiográficos de función ventricular derecha en la hipertensión pulmonar tromboembólica crónica
- 6000-201 - Relación entre el rendimiento y la poscarga pulsátil del ventrículo derecho en pacientes con hipertensión pulmonar tromboembólica crónica
- 6000-198 - Impacto de un programa educacional en pacientes con hipertensión arterial pulmonar en la adherencia al tratamiento con iloprost inhalado
- 6000-204 - Prevalencia de los factores de riesgo para la insuficiencia cardiaca con fracción de eyección preservada del ventrículo izquierdo en la hipertensión arterial pulmonar. Estudio MULTICÉNTRICO
- 6000-349 - Oxigenador de membrana extracorpórea en diferentes formas de insuficiencia cardiaca grave: experiencia en un centro
- 6000-194 - Utilidad de la septostomía auricular como tratamiento paliativo en pacientes con hipertensión pulmonar grave y disfunción ventricular derecha
- 6000-197 - El índice de trabajo sistólico del ventrículo derecho disminuye tras la tromboendarterectomía pulmonar en pacientes con hipertensión pulmonar tromboembólica crónica
-
Jiménez López Guarch, Carmen
- 4008-3 - Hipertensión pulmonar asociada a cardiopatías congénitas y shunt sistémico pulmonar: ¿es siempre mejor cerrar? Datos del Registro Español de Hipertensión Pulmonar (REHAP)
- 6001-402 - Análisis anatómico y funcional del remodelado de cavidades derechas tras cierre percutáneo de defectos del tabique interauricular
- 6000-202 - El índice de trabajo sistólico del ventrículo derecho se correlaciona con los parámetros ecocardiográficos de función ventricular derecha en la hipertensión pulmonar tromboembólica crónica
-
Lázaro Salvador, María
- 4048-12 - Prevención en hipertensión pulmonar. ¿cómo estamos tratando a nuestros pacientes?
- 4008-3 - Hipertensión pulmonar asociada a cardiopatías congénitas y shunt sistémico pulmonar: ¿es siempre mejor cerrar? Datos del Registro Español de Hipertensión Pulmonar (REHAP)
- 4022-8 - Detección de hipertrofia ventricular izquierda por ECG mediante el producto duración por voltaje. Validación por ecocardiografía
- 6000-45 - ¿Cuál es el perfil del paciente con hipertensión pulmonar precapilar en España? Estudio multicéntrico
- 6000-204 - Prevalencia de los factores de riesgo para la insuficiencia cardiaca con fracción de eyección preservada del ventrículo izquierdo en la hipertensión arterial pulmonar. Estudio MULTICÉNTRICO
-
Oliver, José María
- 6001-404 - Terapia de Maze en pacientes con cardiopatía congénita: experiencia de un centro
- 6001-403 - Anomalías del drenaje venoso sistémico y pulmonar en pacientes con síndrome de turner y valvulopatía aórtica y/o afectación aórtica
- 4008-3 - Hipertensión pulmonar asociada a cardiopatías congénitas y shunt sistémico pulmonar: ¿es siempre mejor cerrar? Datos del Registro Español de Hipertensión Pulmonar (REHAP)
- Quero, María
-
Segovia, Javier
- 4028-3 - Miocardiopatía en fibrosis quística: una variante de la enfermedad de Keshan
- 4008-3 - Hipertensión pulmonar asociada a cardiopatías congénitas y shunt sistémico pulmonar: ¿es siempre mejor cerrar? Datos del Registro Español de Hipertensión Pulmonar (REHAP)
- 6001-569 - Seguimiento del corazón trasplantado: evaluación no invasiva y evolución de los parámetros ecocardiográficos en el primer año tras el trasplante cardiaco
- 4049-2 - Insuficiencia cardiaca de alto gasto e hipertensión pulmonar en pacientes con mielofibrosis: descripción de una nueva entidad
- 4045-2 - ANÁLISIS del efecto del everolimus en la progresión de la enfermedad vascular del injerto cardiaco establecida: estudio prospectivo y aleatorizado con ecografía intracoronaria
- 4045-3 - Efecto de la introducción de everolimus en el desarrollo de infecciones, tumores y otras complicaciones en pacientes con enfermedad vascular del injerto: estudio prospectivo y aleatorizado
- 6001-570 - Estudio de deformación miocárdica en la cardiopatía de la fibrosis quística: el valor de los puntos de corte
- 6001-551 - Función y deformación ventricular derecha: ¿la clave para el diagnóstico de rechazo agudo postrasplante?
- 6000-372 - RM cardiaca de estrés con dobutamina en una población de trasplantados cardiacos. Seguridad y viabilidad
- 6000-369 - Evolución ecocardiográfica de los parámetros clásicos y de deformación en la función ventricular derecha durante el primer año postrasplante cardiaco
- 6000-356 - Factores pronóstico en la miocardiopatía dilatada enólica
- 4027-4 - Predictores pronósticos ecocardiográficos en hipertensión pulmonar: más allá de la excursión anular tricúspide
- 4037-6 - Disfunción ventricular izquierda avanzada en fibrosis quística: una nueva forma de miocardiopatía
- 4037-7 - Historia natural de la miocardiopatía dilatada enólica
- 6001-572 - Miocardiopatía asociada a la fibrosis quística: valor de los parámetros ecocardiográficos clásicos y utilidad del strain
- 6000-383 - Evaluación de las presiones de llenado del ventrículo izquierdo para la detección de rechazo agudo en pacientes con trasplante cardiaco
- 6000-270 - ¿Juega algún papel el selenio en la miocardiopatía de la fibrosis quística?
- 6000-320 - Estudio de prevalencia de miocardiopatía en una población de adultos con fibrosis quística
- 4029-7 - Rechazo agudo en el trasplante cardiaco: ¿preparados para reducir el número de procedimientos invasivos? Papel de la ecocardiografía
- 4045-5 - Análisis detallado de eventos clínicos a largo plazo tras la introducción de everolimus en pacientes con enfermedad vascular del injerto establecida: estudio prospectivo y aleatorizado
-
Subirana Domenech, Maite
- 4008-8 - Cierre percutáneo de dehiscencias en las neoaurículas en pacientes con cirugía de Switch auricular
- 6001-394 - Hipertensión pulmonar en adultos con cardiopatía congénita
- 4008-3 - Hipertensión pulmonar asociada a cardiopatías congénitas y shunt sistémico pulmonar: ¿es siempre mejor cerrar? Datos del Registro Español de Hipertensión Pulmonar (REHAP)
- 6001-417 - Accidentes cerebrovasculares de etiología embólica en adultos con tetralogía de fallot. estudio multicéntrico retrospectivo
- 4042-8 - Eplerenona en ventrículo derecho sistémico. ensayo clínico aleatorizado
- 4008-7 - Impacto del embarazo en mujeres con D-transposición de grandes arterias sometidas a switch atrial