ISSN: 0300-8932
Factor de impacto 2024
4,9
SEC 2017 - El Congreso de las Enfermedades Cardiovasculares

Madrid,
26 - 28 de Octubre de 2017
Introducción
Dr. Luis Rodríguez Padial
Presidente del Comité Científico del Congreso
Comité ejecutivo
Comité de evaluadores
Índice de autores
5006. Miocardiopatía: clínica y genética
Fecha
: 26-10-2017 15:15:00
Tipo
: Comunicaciones mini orales
Sala
: Sala Bruselas (Planta 4. Izda.)
5006-5. Características del genotipo y fenotipo de una población de pacientes diagnosticados de miocardiopatía hipertrófica
María Eladia Salar Alcaraz1, Juan Martínez Sánchez2, Inmaculada Pérez Sánchez2, Pablo Peñafiel Verdú2, Esther Guerrero Pérez2, Juan José Santos Mateo2, Arcadio García Alberola2 y Juan Ramón Gimeno Blanes2 del 1Hospital General Universitario Los Arcos del Mar Menor, San Javier (Murcia), y 2Hospital Clínico Universitario Virgen de la Arrixaca, El Palmar (Murcia).
Introducción y objetivos: La miocardiopatía hipertrófica (MCH) MCH se caracteriza por su gran heterogeneidad tanto en la genética como en sus manifestaciones clínicas. El objetivo de este estudio es describir el genotipo y las características clínicas de una población de pacientes diagnosticados de MCH en la consulta de Cardiopatías Familiares.
Métodos: Se seleccionó de manera retrospectiva a los 225 pacientes que se incluyeron en el análisis. Todos eran portadores de una mutación causal. El estudio clínico inicial se basó en la realización de un ECG de 12 derivaciones en reposo, ecocardiograma Doppler y 2D, Holter de 24 horas. También se recogieron los datos disponibles de la ergometría y la RMN cardiaca.
Resultados: Se hallaron 43 mutaciones causales: 24 afectaban a MYBPC3, 15 afectaban a MYH7, 2 afectaban a TNNT2, las 3 mutaciones restantes afectaban a los genes MYL2, MYH6 y FHL1 (figura). Las características clínicas, los resultados de las pruebas complementarias y los eventos en el seguimiento se muestran en la tabla. Los pacientes con mutaciones en el gen MYBPC fueron con más frecuencia varones (73 frente a 54%; p = 0,013), tenían menos síntomas en el momento del diagnóstico, más episodios de TVNS en el Holter (59 frente a 30,8%; p = 0,002), un mayor grosor de la pared del VI (21 ± 5mm frente a 18 ± 6 mm; p = 0,003) y se implantaron más DAI (24 frente a 6%;p = 0,004). Los pacientes con mutaciones en MYBPC3 que provocan truncamiento en la proteína presentaban con más frecuencia TVNS en el Holter (64 frente a 37%; p < 0,001), 3 o más factores de riesgo para MS clásicos (11 frente a 4%; p = 0,047), se les implantó con más frecuencia un DAI (26 frente a 12%; p = 0,001) y tuvieron más eventos arrítmicos en el seguimiento (11 frente a 3%; p = 0,047). También se observaron diferencias al comparar la mutación c.2308+1G > A (n = 53) con las mutaciones p.Arg891Alafs*160 y p.Pro108Alafs*9 (n = 67) en MYBPC3. Los pacientes portadores de la primera mutación tenían una FEVI menor (58 ± 12 frente a 63 ± 10%; p = 0,035), un ECG con alteraciones sugestivas de MCH (74 frente a 48%; p = 0,004), y en el seguimiento presentaron mayor tasa de eventos arrítmicos (19 frente a 4%; p = 0,017) y de eventos combinados (30 frente a 13%; p = 0,025).
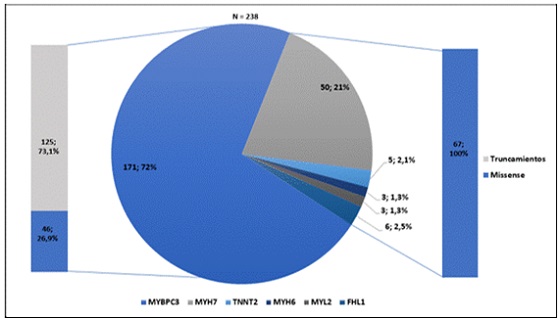
Tipo y distribución de las mutaciones causales halladas en nuestra población.
|
Características clínicas de la población total y analizadas por subgrupos |
||||||||||
|
Características clínicas |
MYBPC3 (n = 162) frente a MYH7 (n = 46) |
p |
Truncamiento (n = 120) frente a no truncamientoa (n = 88) |
p |
c.2308+1G > A frente a p.Arg891Alafs*160 y p.Pro108Alafs*9 |
p |
Todos (n = 225) |
|||
|
Edad, años, media ± desviación estándar |
46,0 ± 15,0 |
49,6 ± 17,2 |
NS |
46,1 ± 14,3 |
45,7 ± 17,2 |
NS |
47,9 ± 14,3 |
44,7 ± 14,3 |
NS |
47,0 ± 15,9 |
|
Sexo masculino, n (%) |
119 (73,5) |
25 (54,3) |
0,01 |
85 (70,8) |
59 (67,0) |
NS |
38 (71,1) |
47 (70,1) |
NS |
152 (67,6) |
|
Dolor torácico, n (%) |
26 (16,0) |
16 (34,8) |
0,005 |
18 (15,0) |
24 (27,3) |
0,02 |
6 (11,3) |
12 (17,9) |
NS |
47 (20,9) |
|
Clase funcional NYHA III-IV, n (%) |
19 (11,7) |
12 (26,1) |
0,02 |
14 (11,7) |
17 (19,3) |
NS |
7 (13,2) |
7 (10,4) |
NS |
32 (14,2) |
|
Síncope previo, n (%) |
27 (16,7) |
9 (19,6) |
NS |
18 (15,0) |
18 (20,5) |
NS |
7 (13,2) |
11 (16,4) |
NS |
40 (17,8) |
|
TVNS en Holter, n (%) |
85 (59,0) |
12 (30,8) |
0,002 |
70 (64,2) |
27 (36,5) |
0,0001 |
35 (70,0) |
35 (59,3) |
NS |
103 (52,3) |
|
ECG normal, n (%) |
12 (7,4) |
3 (6,1) |
NS |
9 (7,5) |
6 (6,8) |
NS |
2 (3,8) |
7 (10,4) |
NS |
16 (7,1) |
|
ECG sugestivo, n (%) |
100 (61,7) |
31 (63,3) |
NS |
71 (59,2) |
57 (64,8) |
NS |
39 (73,6) |
32 (47,8) |
0,004 |
136 (60,4) |
|
Grosor máximo de la pared, mm |
21,0 ± 5,4 |
18,3 ± 5,5 |
0,03 |
20,8 ± 5,5 |
20,4 ± 5,4 |
NS |
21,1 ± 5,7 |
20,6 ± 5,4 |
NS |
20,0 ± 5,6 |
|
Obstrucción en el TSVI, n (%) |
45 (27,8) |
17 (37,0) |
NS |
33 (27,5) |
29 (33) |
NS |
12 (22,6) |
21 (31,3) |
NS |
65 (28,9) |
|
FEVI < 50%, n (%) |
16 (9,9) |
5 (10,9) |
NS |
13 (10,8) |
8 (9,1) |
NS |
10 (18,9) |
3 (4,5) |
0,012 |
23 (10,2) |
|
Implante de DAI, n (%) |
39 (24,1) |
3 (6,5) |
0,004 |
31 (25,8) |
13 (12,4) |
0,011 |
18 (34) |
13 (19,4) |
NS |
44 (19,5) |
|
Muerte súbita, n (%) |
4 (2,5) |
1 (2,2) |
NS |
3 (2,5) |
2 (2,3) |
NS |
6 (11,3) |
2 (3,0) |
NS |
3 (1,3) |
|
Muerte por IC, n (%) |
3 (1,9) |
1 (2,2) |
NS |
3 (2,5) |
1 (1,1) |
NS |
3 (5,7) |
0 |
NS |
4 (1,8) |
|
Descarga DAI, n (%) |
8 (4,9) |
0 |
NS |
8 (6,7) |
0 |
0,008 |
6 (11,3) |
2 (3,0) |
NS |
8 (3,6) |
|
Ingreso por IC, n (%) |
14 (8,6) |
5 (10,9) |
NS |
11 (9,2) |
8 (9,1) |
NS |
5 (9,4) |
6 (9,0) |
NS |
21 (9,3) |
|
Trasplante cardiaco, n (%) |
5 (3,1) |
0 |
NS |
4 (3,3) |
1 (1,1) |
NS |
2 (3,8) |
2 (3,0) |
NS |
7 (3,1) |
|
Eventos arrítmicos mayoresb, n (%) |
15 (9,3) |
1 (2,2) |
NS |
13 (10,8) |
3 (3,4) |
0,047 |
10 (18,9) |
3 (4,5) |
0,017 |
17 (7,6) |
|
Eventos cardiovasculares combinadosc, n (%) |
30 (18,5) |
6 (13,0) |
NS |
25 (20,8) |
11 (12,5) |
NS |
16 (30,2) |
9 (13,4) |
0,04 |
38 (16,9) |
|
aSe excluyeron del análisis pacientes con mutaciones en otros genes (no MYH7) y con mutaciones dobles (n = 13). bIncluye: MS, TVMS, descarga de DAI. cIncluye: eventos arrítmicos mayores, ingreso por IC, trasplante cardiaco y muerte por IC. |
||||||||||
Conclusiones: Las mutaciones más frecuentes en nuestro medio afectan al gen MYBPC3. Las 3 más prevalentes provocan un truncamiento en la proteína sintetizada. Estas mutaciones se asocian a un peor pronóstico, siendo c.2308+1G > A la mutación más agresiva en esta serie.
Comunicaciones disponibles de "Miocardiopatía: clínica y genética"
- 5006-1. Presentación
- Juan Ramón Gimeno Blanes, Murcia, y Gonzalo Barge Caballero, A Coruña.
- 5006-2. Valor pronóstico del estudio de la función auricular izquierda en pacientes con miocardiopatía hipertrófica mediante resonancia magnética cardiaca tissue tracking
- Rocío Hinojar1, Covadonga Fernández-Golfín1, María Ángeles Fernández-Méndez2, Amparo Esteban2, María Plaza-Martín1, Ariana González-Gómez1, Luis Miguel Rincón1 y José Luis Zamorano1 del 1Servicio de Cardiología y 2Servicio de Radiología, Hospital Universitario Ramón y Cajal, Madrid.
- 5006-3. Miocardiopatía de tako-tsubo y edema intramiocárdico: distribución regional y tendencia temporal de desaparición del edema en fase aguda
- Paula Antuña Álvarez, Jorge Salamanca, Teresa Alvarado, Alberto Cecconi, Eduardo Pozo, Río Aguilar, Luis Jesús Jiménez-Borreguero y Fernando Alfonso del Hospital Universitario de La Princesa, Madrid.
- 5006-4. Tratamiento con flecainida de una mutación en SCN5A asociada a miocardiopatía dilatada y arritmias ventriculares
- Mercedes Cabrera Ramos, Juan Jiménez Jáimez, Francisco José Bermúdez Jiménez, Laura Pertejo Manzano, Lorena González Camacho, Isabel Gallardo Sánchez, Miguel Álvarez López y Luis Tercedor Sánchez del Hospital Universitario Virgen de las Nieves, Granada.
- 5006-5. Características del genotipo y fenotipo de una población de pacientes diagnosticados de miocardiopatía hipertrófica
- María Eladia Salar Alcaraz1, Juan Martínez Sánchez2, Inmaculada Pérez Sánchez2, Pablo Peñafiel Verdú2, Esther Guerrero Pérez2, Juan José Santos Mateo2, Arcadio García Alberola2 y Juan Ramón Gimeno Blanes2 del 1Hospital General Universitario Los Arcos del Mar Menor, San Javier (Murcia), y 2Hospital Clínico Universitario Virgen de la Arrixaca, El Palmar (Murcia).
- 5006-6. Implicación pronóstica del tratamiento farmacológico al alta en el síndrome de Tako-Tsubo. Seguimiento a muy largo plazo del registro RETAKO
- Manuel Almendro Delia1, Beatriz Lorenzo López1, Iván Javier Núñez Gil2, Mireia Andrés3, Alessandro Sionis4, Ana Martín5, Teresa Bastante6 y Juan Carlos García Rubira. En Representación de los Investigadores del Registro RETAKO1 del 1Hospital Universitario Virgen Macarena, Sevilla, 2Hospital Clínico San Carlos, Madrid, 3Hospital Universitario Vall d';Hebron, Barcelona, 4Hospital de Sant Pau, Barcelona, 5Hospital Universitario de Salamanca y 6Hospital Universitario de La Princesa, Madrid.
- 5006-7. Prevalencia y rentabilidad diagnóstica comparadas del cribado en las cardiopatías hereditarias. Registro 10K
- David López Cuenca, Elisa Nicolás, Antonio Pastor, Marina Navarro Peñalver, Inmaculada Pérez Sánchez, Carmen Muñoz Esparza, María Sabater y Juan Ramón Gimeno-Blanes del Hospital Clínico Universitario Virgen de la Arrixaca, El Palmar (Murcia).
- 5006-8. Genética en miocardiopatía dilatada familiar y truncamientos en titina
- Alba Sádaba Cipriaín, Carolina Tiraplegui Garjón, Gemma Lacuey Lecumberri, Maite Basurte Elorz, Isabel Santos Sánchez, Lorena Malagón López, Pablo Bazal Chacón e Ignacio Roy Añon del Complejo Hospitalario de Navarra, Pamplona (Navarra).
Más comunicaciones de los autores
-
García Alberola, Arcadio
- 6002-44 - Correlación entre el riesgo arrítmico estimado por el modelo HCM-Risk-SCD y los eventos arrítmicos observados en una población de pacientes con miocardiopatía hipertrófica
- 6003-61 - Discordancia en la estratificación del riesgo según las escalas de riesgo en los pacientes con fibrilación auricular no valvular que inician anticoagulantes orales directos
- 5023-6 - Efectividad y seguridad de los anticoagulantes orales directos en pacientes con fibrilación auricular no valvular en práctica clínica real
- 6002-43 - Evolución de la función ventricular y terapias apropiadas tras implante de desfibrilador automático implantable en prevención primaria. Implicaciones de cara al recambio del dispositivo
- 5021-8 - Predictores de mortalidad en pacientes con fibrilación auricular no valvular que inician anticoagulantes orales directos
- 5023-4 - Cumplimiento de las recomendaciones sobre monitorización de la función renal tras la prescripción de anticoagulantes orales no antagonistas de la vitamina K en pacientes con fibrilación auricular
- 5023-3 - Determinantes clínicos de dosificación inapropiada al iniciar anticoagulantes orales directos en pacientes con fibrilación auricular no valvular
- 6012-207 - Patrones electrocardiográficos de una población de pacientes con miocardiopatía hipertrófica
- 6003-62 - Comparación de las escalas de riesgo trombótica y hemorrágicas en pacientes con fibrilación auricular no valvular que inician tratamiento con anticoagulantes orales directos
- 5006-5 - Características del genotipo y fenotipo de una población de pacientes diagnosticados de miocardiopatía hipertrófica
- 7005-7 - Experiencia de seguimiento de pacientes con marcapasos desde un hospital comarcal
-
Gimeno Blanes, Juan Ramón
- 5006-5 - Características del genotipo y fenotipo de una población de pacientes diagnosticados de miocardiopatía hipertrófica
- 5006-1 - Presentación
- 7002-18 - Espectro genético de la miocardiopatía arritmogénica causante de muerte súbita
- 6002-44 - Correlación entre el riesgo arrítmico estimado por el modelo HCM-Risk-SCD y los eventos arrítmicos observados en una población de pacientes con miocardiopatía hipertrófica
- 5026-5 - Incidencia y pronóstico a largo plazo de la miocardiopatía de Tako-Tsubo en una cohorte de 65 pacientes consecutivos
- 6012-199 - Análisis de las muertes súbitas por miocardiopatía arritmogénica en nuestro entorno
- 5006-7 - Prevalencia y rentabilidad diagnóstica comparadas del cribado en las cardiopatías hereditarias. Registro 10K
- 7009-9 - Resultado inmediato y a medio plazo de la estenosis aórtica grave tratada mediante implante prótesis percutánea Sapien 3 en pacientes de alto riesgo
- 6012-196 - Heterogeneidad en la activación eléctrica en la miocardiopatía arritmogénica a partir del vectocardiograma y el análisis de componentes principales del electrocardiograma
- 6012-196 - Heterogeneidad en la activación eléctrica en la miocardiopatía arritmogénica a partir del vectocardiograma y el análisis de componentes principales del electrocardiograma
- 6012-207 - Patrones electrocardiográficos de una población de pacientes con miocardiopatía hipertrófica
-
Guerrero Pérez, Esther
- 6034-422 - Pronóstico de los pacientes con insuficiencia renal avanzada previa al implante de prótesis aórtica percutánea
- 7007-4 - Análisis y diferencias en mortalidad entre los pacientes con infarto de miocardio tipo 1 y tipo 2
- 6048-568 - Evaluación del impacto del número de fármacos recomendados al alta en la mortalidad al año tras un síndrome coronario agudo
- 5006-5 - Características del genotipo y fenotipo de una población de pacientes diagnosticados de miocardiopatía hipertrófica
- 5026-3 - Bloqueo completo de rama derecha como valor pronóstico adicional sobre la escala GRACE para la predicción de mortalidad hospitalaria en pacientes con síndrome coronario agudo
- 6012-207 - Patrones electrocardiográficos de una población de pacientes con miocardiopatía hipertrófica
-
Martínez Sánchez, Juan
- 5006-5 - Características del genotipo y fenotipo de una población de pacientes diagnosticados de miocardiopatía hipertrófica
- 6002-43 - Evolución de la función ventricular y terapias apropiadas tras implante de desfibrilador automático implantable en prevención primaria. Implicaciones de cara al recambio del dispositivo
- 6012-207 - Patrones electrocardiográficos de una población de pacientes con miocardiopatía hipertrófica
-
Peñafiel Verdú, Pablo
- 6002-43 - Evolución de la función ventricular y terapias apropiadas tras implante de desfibrilador automático implantable en prevención primaria. Implicaciones de cara al recambio del dispositivo
- 5006-5 - Características del genotipo y fenotipo de una población de pacientes diagnosticados de miocardiopatía hipertrófica
- 6012-207 - Patrones electrocardiográficos de una población de pacientes con miocardiopatía hipertrófica
- 6008-129 - Nueva escala para el diagnóstico de seguridad de las taquicardias regulares de QRS ancho
- Pérez Sánchez, Inmaculada
-
Salar Alcaraz, María Eladia
- 6053-632 - Educación sanitaria y cuidados de enfermería en pacientes con desfibrilador automático implantable en la fase aguda
- 5006-5 - Características del genotipo y fenotipo de una población de pacientes diagnosticados de miocardiopatía hipertrófica
- 6012-207 - Patrones electrocardiográficos de una población de pacientes con miocardiopatía hipertrófica
-
Santos Mateo, Juan José
- 6004-89 - Metformina previene la fibrosis cardiaca tras infarto de miocardio, a través de la atenuación de la ruta de señalización NADPH oxidasa/PKCa/Gal-3
- 6028-365 - Utilidad de la gammagrafía con Tc99m-DPD en el diagnóstico de amiloidosis cardiaca
- 6002-43 - Evolución de la función ventricular y terapias apropiadas tras implante de desfibrilador automático implantable en prevención primaria. Implicaciones de cara al recambio del dispositivo
- 6041-500 - Tratamiento con diálisis peritoneal continua ambulatoria en pacientes con insuficiencia cardiaca avanzada refractaria a tratamiento diurético
- 5006-5 - Características del genotipo y fenotipo de una población de pacientes diagnosticados de miocardiopatía hipertrófica
- 6041-526 - Prevalencia, predictores e impacto pronóstico del deterioro agudo de la función renal (criterios RIFLE) durante la hospitalización por insuficiencia cardiaca descompensada
- 5031-3 - Prevalencia de amiloidosis senil por captación miocárdica de Tc99m-DPD en una población de 1.501 gammagrafías óseas consecutivas no seleccionadas
- 6002-44 - Correlación entre el riesgo arrítmico estimado por el modelo HCM-Risk-SCD y los eventos arrítmicos observados en una población de pacientes con miocardiopatía hipertrófica
- 6012-207 - Patrones electrocardiográficos de una población de pacientes con miocardiopatía hipertrófica